
A 2026 Nature Neuroscience study from the Igarashi lab proposes a circuit-level answer for why the lateral entorhinal cortex (LEC) is the first cortical region to falter in Alzheimer’s: dopamine inputs from the VTA and substantia nigra to the LEC go functionally silent at the earliest disease stage in an APP knock-in mouse — before any axonal degeneration — and L-DOPA reverses the resulting memory deficit.1
Research Highlights
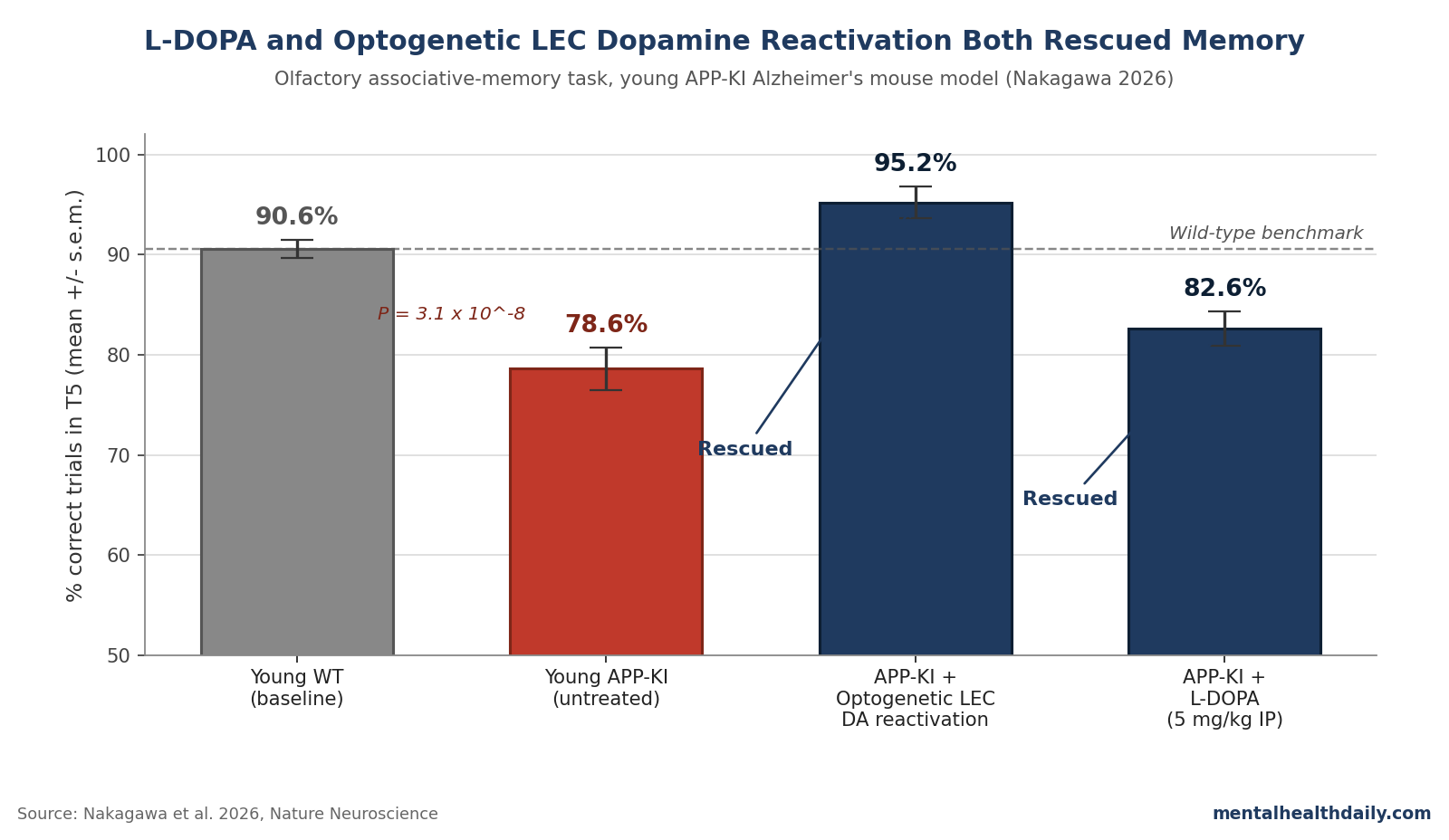
- Nakagawa 2026 recorded LEC neurons and VTA/SNc–to–LEC dopamine fibers in APP knock-in (APP-KI) mice using a previously validated olfactory associative-memory task. Young APP-KI mice (3–6 months) reached only 78.6% correct trials in the late-trial window vs. 90.6% for wild-type, with the gap widening in older animals.1
- Tyrosine hydroxylase–positive (TH+) dopamine fibers in the LEC were not significantly reduced in young APP-KI mice. The wiring is there; the release is not. Dopamine signals for novel rewarded cues at the earliest learning timepoint were specifically blunted (P = 4.2 × 10−3).1
- Optogenetic reactivation of LEC dopamine fibers during novel-cue trials rescued performance from 77.9% to 95.2% correct in T5 and the share of fully-learned sessions from 50.1% to 86.9%.1
- Intraperitoneal L-DOPA (5 mg/kg/day for 5 days) restored memory in APP-KI mice (67.1% → 82.6% correct, P = 1.1 × 10−3) and also rescued the PS19 tau model (64.8% → 84.8%). L-DOPA had no effect on healthy wild-type controls.1
- The headline is a mechanism, not a treatment. No human trial has yet measured LEC dopamine in patients with AD, and the existing dopaminergic-AD clinical work (rotigotine, levodopa add-on) is small and mixed.4,5
Translation boundary: this is a mouse finding, not a clinical trial. The mechanism it sketches — intact dopamine wiring with broken release for novel rewarded cues — is testable in human brains.
It also lines up with a small clinical literature suggesting dopaminergic agents might do something in early AD that cholinesterase inhibitors don’t.2,3
Popular coverage of an early-AD mouse paper defaults to two failure modes: framing it as “Alzheimer’s rescue” (it isn’t), or burying it as “another mouse study” (which underweights an unusually clean structural-vs.-functional dissociation). The more useful read: this is a mechanism finding pointing to a druggable target that’s already on every neurology formulary.
Why the LEC Fails First in Alzheimer’s — and Why Dopamine Is the Suspect
The lateral entorhinal cortex sits at the top of the hippocampal–entorhinal memory circuit and shows the earliest measurable functional decline in preclinical AD on fMRI — earlier than the medial entorhinal cortex and earlier than the hippocampus itself.6 Braak and Braak placed entorhinal layer 2 neurons among the first cortical casualties of tau pathology,7 and profound loss of these neurons has been documented in postmortem brains of patients diagnosed with very mild AD.8
What hasn’t been clear is the mechanism of selective LEC vulnerability. Nakagawa adds a circuit-level candidate: the LEC is densely innervated by VTA and SNc dopamine projections, dopamine neurons are intrinsically vulnerable to oxidative stress (the same vulnerability that drives Parkinson’s), and LEC dopamine is needed for encoding novel cue–reward associations even in healthy mice.9
The hippocampus, by contrast, receives little direct VTA/SNc input — its dopamine arrives largely via the locus coeruleus, which co-releases norepinephrine.10 That circuit asymmetry between LEC and hippocampus is the lever Nakagawa exploits.
APP-KI Mice Failed at Novel Associations but Kept Old Ones
The APP knock-in line (APPNL-G-F/NL-G-F) is a Saito-lab construct that produces gradual Aβ accumulation without the protein-overexpression artifacts of older transgenic AD mice.11 Aβ deposition in the LEC begins around 2 months of age in this line.
The task — established in the Igarashi lab’s 2021 Nature paper — trains head-fixed mice to lick for sucrose after a rewarded odor (Odor-A) and withhold licking after a punished odor (Odor-B), then introduces novel odor pairs on top of the prelearned ones.9
The dissociation in the APP-KI mice was sharp: prelearned Odor-A/B performance was indistinguishable from wild-type across both age groups (P = 0.92 in young, P = 0.61 in old, post hoc Tukey). Novel-association performance was impaired.
Young APP-KI mice reached 78.6 ± 2.1% correct trials in the final third of the session (T5) vs. 90.6 ± 0.9% in young wild-types (F1,50 = 20.2, P = 4.1 × 10−5; n = 11 WT and 16 APP-KI).1
The session-level signal was even larger. APP-KI mice reached the ≥80% “correct session” criterion only 59.1% of the time vs. 77.5% in wild-type at the young age, and 47.3% vs. 81.3% at 7–11 months — a near-halving of correct sessions in older APP-KI animals (P = 1.3 × 10−3).1
Translation: the deficit is not memory in general. It is the specific ability to bind a novel rewarded cue to its outcome — which is what the LEC is for.
LEC Dopamine Wiring Was Intact — Release for Novel Cues Was Not
The structural-vs.-functional dissociation is the key result. Anti-TH immunostaining of dopamine fibers in the LEC of APP-KI mice showed only a non-significant trend toward decreased density (F1,24 = 0.79 for strain, P = 0.38) at both 3–6 and 7–11 months.
Cell-body counts of dopaminergic neurons in the VTA and SNc were also unchanged.1 The wiring is preserved.
Dopamine release failed before structure did. Fiber photometry of GCaMP-expressing dopamine fibers in the LEC of APP-KI × DAT-Cre mice showed that dopamine release for novel rewarded cues (Odor-1) was significantly blunted at the earliest learning timepoint (T1) compared to wild-type controls.
The model showed a strain × time interaction (F4,80 = 2.5, P = 0.047), with a T1-specific gap across all sessions (P = 4.2 × 10−3) and correct sessions (P = 4.8 × 10−3).1 Dopamine signals for the prelearned Odor-A were comparable between groups.
The pattern is striking: synaptic terminals are present, dopamine reserves appear adequate (the optogenetic rescue confirms this), but the release machinery fails specifically for the novel-cue condition that demands it most. Whatever Aβ is doing in the LEC, it disables release before it kills the fiber.
Optogenetic and L-DOPA Rescue Both Worked — the L-DOPA Result Is the Translational One
Two interventions restored LEC associative-memory function in young APP-KI mice. The optogenetic rescue is the cleaner mechanism test. The L-DOPA rescue is the one that maps onto a drug humans can take.
- Optogenetic reactivation. ChR2 was expressed in dopamine fibers of APP-KI × DAT-Cre mice and the LEC was photo-stimulated only during novel Odor-1 trials. Late-trial correct rate rose from 77.9% to 95.2% (P = 0.024). Correct-session rate rose from 50.1% to 86.9% (P = 0.019). Stimulating across all four odor types didn’t help — the deficit is selective to novel-cue trials, and the rescue must be too.1
- L-DOPA (oral dopamine precursor). Intraperitoneal L-DOPA (5 mg/kg in saline, 30 minutes before behavior, 5 consecutive days) raised T5 performance in young APP-KI mice from 67.1% to 82.6% (F1,30 = 5.2, P = 0.029; post hoc P = 1.1 × 10−3). Correct-session rate climbed from 33.1% to 78.5% (P = 7.0 × 10−4). The same dose also rescued associative learning in the PS19 transgenic tau model of AD (64.8% → 84.8%, P = 4.7 × 10−3). L-DOPA had no effect on wild-type controls — consistent with a deficit-correction action rather than a generalized cognitive enhancer.1
The PS19 result extends the mechanism beyond amyloid. PS19 mice carry a mutant human tau transgene and develop tau pathology rather than amyloid pathology — a distinct disease mechanism from APP-KI. The fact that L-DOPA rescues both points to LEC dopamine dysfunction as a downstream consequence of multiple AD-relevant pathologies, not a peculiarity of the APP-KI line.
Intact Mouse Dopamine Wiring Does Not Prove Human Alzheimer’s Reversibility
Mouse-model translation has a brutal track record in AD — semagacestat, tarenflurbil, IVIG, and multiple amyloid antibodies before the partial wins of lecanemab and donanemab.12 Three limits keep the Nakagawa result in mechanism territory.
Healthy reserve in mice exceeds that in elderly humans with AD. Young APP-KI mice have intact dopamine cell counts and intact LEC fiber density — only function is impaired.
By the time a human is symptomatic for AD, both VTA dopamine cell counts and entorhinal layer 2 neuron counts are reduced.8,13 Restoring release in a mouse with intact wiring is a different problem from restoring it in a human with degenerated wiring.
L-DOPA in AD patients is not a fresh hypothesis. Koch et al. randomized 94 patients with mild-to-moderate AD to 24 weeks of the D1/D2/D3 agonist rotigotine vs. placebo and reported preserved frontal-network function and stable activities of daily living in the rotigotine arm, though primary cognitive endpoints didn’t separate.4
Earlier acute-L-DOPA work showed transient modulation of cortical cholinergic transmission in AD patients without sustained cognitive benefit.5 Nakagawa gives those mixed results a sharper hypothesis: target early-stage patients, measure LEC-dependent novel associative memory specifically, and stop expecting global cognition to move.
No one has measured LEC dopamine in human AD brains. Dopamine fibers exist in the human entorhinal cortex,14 but no PET tracer or postmortem study has yet quantified entorhinal dopamine in patients with AD. The translational pathway suggested here doesn’t exist as a clinical study yet.
LEC Dopamine Dysfunction Sharpens Older Dopamine Findings in AD
The dopamine-in-AD literature is older and stranger than most readers think. Postmortem and PET studies have documented decreased D2 receptor expression in the hippocampus and reduced striatal dopamine transporter binding in some AD cohorts.2 Pan’s network meta-analysis of dopamine-system biomarkers found small-to-moderate reductions in receptor binding across multiple regions, with high heterogeneity across studies.3
The dominant interpretation has treated this as incidental — a downstream casualty rather than a mechanism. Nakagawa flips that: dopamine dysfunction in the LEC specifically is the proximal cause of the earliest memory symptoms, and the wider striatal/hippocampal dopamine changes documented in postmortem studies are downstream. The rotigotine trial provides the closest existing anchor for testing this in humans.4
Limitations of the Nakagawa Synthesis
Mouse-only. Every behavioral, photometric, and rescue result is in mice. The translational chain from APP-KI mouse to human AD has historically failed at the bedside, and mechanism elegance in mice is not predictive of clinical efficacy.12
One task, one paradigm. The olfactory cue–outcome task is exquisitely LEC-dependent and was developed by the same lab. It maps cleanly onto novel-association memory but doesn’t cover spatial, working, or episodic recall.
The mechanism upstream of release failure is unspecified. The paper shows release is broken; it doesn’t show why. Whether the failure is presynaptic Aβ toxicity, altered firing at the cell body, or something else upstream is left for future work.
L-DOPA mechanism in AD remains under-specified. L-DOPA raises dopamine globally across multiple dopamine pathways. The behavioral rescue could in principle be mediated by striatal effects on motivation or reinforcement learning rather than LEC release per se.
The optogenetic rescue addresses this only in part — it shows LEC dopamine is sufficient for rescue, not that L-DOPA’s mechanism is necessarily LEC-mediated.
Dose and duration are short. 5 mg/kg/day for 5 days in mice does not extrapolate to chronic human dosing. L-DOPA in humans loses efficacy over years in Parkinson’s and induces dyskinesias; long-term tolerability in non-Parkinsonian AD patients is essentially unknown.
What Researchers Ask About L-DOPA and the Entorhinal Cortex in Alzheimer’s
Does this mean L-DOPA treats Alzheimer’s?
No. It means L-DOPA rescued a specific memory deficit in two AD-relevant mouse models. There is no human trial showing L-DOPA reverses or slows clinical AD. The closest human evidence is a 24-week trial of the dopamine agonist rotigotine in mild-to-moderate AD, which preserved some functional measures but did not separate on the primary cognitive endpoint.4
Why the lateral entorhinal cortex specifically?
The LEC is the cortical area where AD pathology and functional decline appear earliest, before symptoms.6,7 It receives unusually dense dopamine projections from the VTA and SNc, and these projections are required for encoding novel cue–reward associations in healthy mice.9 The Nakagawa paper argues this circuit’s reliance on vulnerable dopamine neurons explains why the LEC fails first.
Did the dopamine fibers actually die in these mice?
Not at the early disease stage tested. TH+ fiber density in the LEC and TH+ cell counts in the VTA and SNc were not significantly reduced in young APP-KI mice — the wiring was intact, but release was selectively impaired for novel rewarded cues. This is part of why optogenetic and L-DOPA rescue worked: the dopamine reserves were available to release.1
How is APP-KI different from older AD mouse models?
APPNL-G-F mice produce Aβ at endogenous physiological levels via humanized knock-in mutations, rather than overexpressing APP with a transgene. They develop Aβ pathology more gradually, without the overexpression artifacts of older models. They don’t develop tau tangles, so the PS19 tau-model rescue in the same paper functions as a complementary test.11
Should patients with mild cognitive impairment ask their doctor for L-DOPA?
No. L-DOPA carries dose-dependent risks (impulse-control disorders, dyskinesia, orthostatic hypotension) characterized in Parkinson’s populations but not in non-Parkinsonian AD patients. This is a mechanism finding that argues for a clinical trial, not for off-label prescription.
What would the right human follow-up look like?
A trial in early-stage AD or amnestic MCI, using a novel-association memory task sensitive to LEC function as the primary endpoint, ideally paired with a dopamine PET tracer measuring entorhinal dopamine. The rotigotine trial is the closest existing precedent.4
References
- Early dopamine disruption in the entorhinal cortex of a knock-in model of Alzheimer’s disease. Nakagawa T, Xie JL, Park K, et al. Nature Neuroscience. 2026. doi:10.1038/s41593-026-02260-w
- Is dopamine involved in Alzheimer’s disease? Martorana A, Koch G. Frontiers in Aging Neuroscience. 2014;6:252. doi:10.3389/fnagi.2014.00252
- Dopamine and dopamine receptors in Alzheimer’s disease: a systematic review and network meta-analysis. Pan X, Kaminga AC, Wen SW, et al. Frontiers in Aging Neuroscience. 2019;11:175. doi:10.3389/fnagi.2019.00175
- Effect of rotigotine vs placebo on cognitive functions among patients with mild to moderate Alzheimer disease: a randomized clinical trial. Koch G, Motta C, Bonnì S, et al. JAMA Network Open. 2020;3(7):e2010372. doi:10.1001/jamanetworkopen.2020.10372
- Dopamine modulates cholinergic cortical excitability in Alzheimer’s disease patients. Martorana A, Mori F, Esposito Z, et al. Neuropsychopharmacology. 2009;34(10):2323–2328. doi:10.1038/npp.2009.60
- Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Khan UA, Liu L, Provenzano FA, et al. Nature Neuroscience. 2014;17(2):304–311. doi:10.1038/nn.3606
- Neuropathological stageing of Alzheimer-related changes. Braak H, Braak E. Acta Neuropathologica. 1991;82(4):239–259. doi:10.1007/bf00308809
- Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. Gomez-Isla T, Price JL, McKeel DW Jr, et al. Journal of Neuroscience. 1996;16(14):4491–4500. doi:10.1523/jneurosci.16-14-04491.1996
- Dopamine facilitates associative memory encoding in the entorhinal cortex. Lee JY, Jun H, Soma S, et al. Nature. 2021;598(7880):321–326. doi:10.1038/s41586-021-03948-8
- Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Kempadoo KA, Mosharov EV, Choi SJ, Sulzer D, Kandel ER. Proceedings of the National Academy of Sciences. 2016;113(51):14835–14840. doi:10.1073/pnas.1616515114
- Single App knock-in mouse models of Alzheimer’s disease. Saito T, Matsuba Y, Mihira N, et al. Nature Neuroscience. 2014;17(5):661–663. doi:10.1038/nn.3697
- Alzheimer’s disease drug development pipeline: 2023. Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2023;9(2):e12385. doi:10.1002/trc2.12385
- Selective neuronal vulnerability in Parkinson’s disease. Gonzalez-Rodriguez P, Zampese E, Surmeier DJ. Progress in Brain Research. 2020;252:61–89. doi:10.1016/bs.pbr.2020.02.005
- The distribution of tyrosine hydroxylase-immunoreactive fibers in the human entorhinal cortex. Akil M, Lewis DA. Neuroscience. 1994;60(4):857–874. doi:10.1016/0306-4522(94)90268-2