
“Schizophrenia ages you faster” is the headline that gets repeated whenever a new biomarker study lands. The 2026 Fernandez-Egea, Garcia-Rizo and Kirkpatrick review — covering 170 studies across mortality, brain imaging, telomeres, epigenetic clocks, and metabolic markers — pushes back on that framing.2
The aging signal is genuine. “Accelerated” is the part that overshoots for most biological systems measured after illness onset. But the review’s own evidence — and its own discussion — contains a more uncomfortable possibility: that both the illness and the aging phenotype may reflect the same upstream developmental damage, making the question of which caused which harder to answer than the headline suggests.
Research Highlights
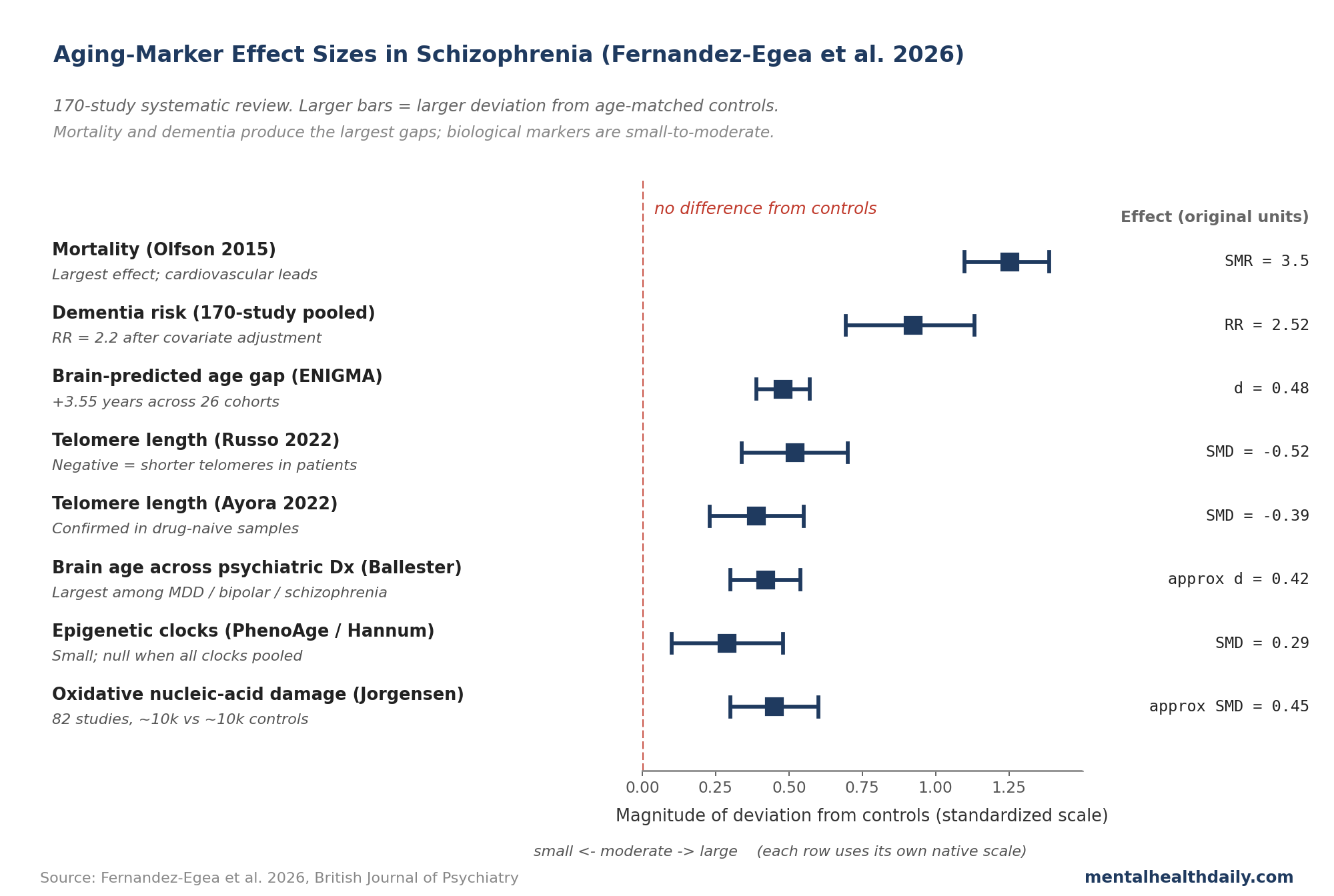
- People with schizophrenia die 15–20 years earlier on average (SMR 2.5–3.5x), develop dementia roughly 20 years earlier, and show metabolic disease in their 20s and 30s. The 2008 “accelerated aging” hypothesis was coined to explain that pattern.1,6,7
- A 2026 systematic review of 170 studies argues “advanced aging” fits better than “accelerated aging,” with an early level-shift followed by parallel decline rather than a steeper slope. The verdict only holds from first episode onward — clinical-high-risk individuals already show brain-age deviations of +1.4 to +6.9 years before diagnosis, consistent with pre-onset accumulation the review cannot rule out.2
- The same authors propose that early life stress may be a common antecedent for both schizophrenia and the aging phenotype — meaning both may reflect shared developmental damage rather than the illness causing the aging.2
- Telomeres run shorter (SMD −0.39 to −0.52) and brain-predicted age is +3.5 years on 26 ENIGMA cohorts (95% CI 2.91–4.19), with most metabolic and inflammatory abnormalities showing up in drug-naïve first-episode patients — meaning antipsychotics did not create the phenotype, though the review concedes they amplify specific subdomains.2,3,4
- The “advanced not accelerated” verdict has three structural problems: biomarker tools are calibrated on healthy people and may miss aging processes they were not trained to detect; the parallel-trajectory finding sidesteps mortality, the most direct aging endpoint, which is not a surrogate; and adjusted estimates likely understate the true effect because studies control for obesity and smoking as if they were confounders rather than mediators of the aging signal.2
Evaluating “Accelerated Aging” in Schizophrenia
The original 2008 paper by Kirkpatrick and colleagues coined “schizophrenia as a syndrome of accelerated aging” to make sense of an awkward observation: people with schizophrenia die 15–20 years earlier than the general population, develop dementia at higher rates, and show metabolic disease in their 20s and 30s.1 If the brain disorder were the whole story, the body shouldn’t be falling apart that fast.
The hypothesis: schizophrenia is a multi-system condition in which biological aging runs ahead of chronological aging across organs. Telomeres should be shorter. Inflammatory and oxidative-stress markers should be elevated. Brain volume and cognition should look like older people’s. Mortality from age-linked diseases should arrive earlier.
“Accelerated” carried a specific implication: a steeper slope. If aging is accelerated in schizophrenia, the gap between patients and matched controls should widen with time. That is the prediction the 2026 review tested against 15 years of accumulated data.
Mortality and Dementia Risk in Schizophrenia: What the Numbers Show
Two findings support the original hypothesis cleanly.
- Mortality: an Olfson 2015 meta-analysis of US national data found a standardized mortality ratio of 3.5 in adults with schizophrenia, cardiovascular disease leading.6 Saha’s earlier global meta-analysis of 37 studies put the all-cause SMR at 2.58.7 The 15–20 year life-expectancy gap holds across countries with very different healthcare systems, ruling out a pure access story.
- Dementia: the 2026 review’s pooled analysis of 11 cohort studies found a relative risk of 2.52 for dementia in non-affective psychosis, holding at 2.2 after adjusting for cardiovascular risk factors and antipsychotic exposure.2 A Danish national registry put dementia diagnosis at 22% by age 65 in schizophrenia versus 6% in the general population.2 The age curve is shifted earlier by roughly 20 years.
Both findings survive covariate adjustment. Both are detectable in first-episode samples before chronic antipsychotic exposure. These are the strongest pillars under the aging-disorder framing.
Brain Age, Telomeres, and Epigenetic Clocks in Schizophrenia
The three biological markers with the most data show consistent deviations, though smaller than the popular framing suggests.
- Brain-predicted age. Machine-learning models trained on healthy MRI scans estimate a “brain age” from structural images. The ENIGMA Schizophrenia Working Group pooled 26 international cohorts (2,803 patients, 2,598 controls) and found a brain-predicted age difference of +3.55 years in schizophrenia (95% CI 2.91–4.19), with a moderate effect size (Cohen’s d = 0.48 — patients’ brains look about half a standard deviation older than chronological age would predict).4 Across psychiatric disorders, schizophrenia produces the largest brain-age deviation (+3.08 years), followed by bipolar (+1.93) and major depression (+1.12).5 The gap shows up in first-episode and clinical-high-risk samples, ranging from +1.4 to +6.9 years.2
- Telomere length. Telomeres are protective caps at the ends of chromosomes that shorten with cell division and chronic stress; shorter telomeres are a standard cellular-aging marker. Four meta-analyses converge on a small-to-medium effect — SMD from −0.388 (Ayora 2022, 22 studies, 4,145 patients vs. 4,184 controls) to −0.52 (Russo 2022, 18 studies).3,2 The shortening shows up in antipsychotic-naïve first-episode samples, so treatment isn’t the only driver.
- Epigenetic clocks. DNA-methylation patterns predict biological age, and several “clocks” exist (Horvath, Hannum, PhenoAge, GrimAge, DunedinPACE). A 2022 meta-analysis found no overall acceleration when clocks were pooled together.9 Stratifying by clock type changes the picture: PhenoAge and Hannum showed small acceleration (SMD around 0.29), GrimAge accelerations of up to 5 years largely disappeared after smoking adjustment, and at-risk patients sometimes look decelerated on Horvath’s clock — epigenetically younger than chronological age, possibly reflecting maturational delay. Higgins-Chen and colleagues clarified why different clocks disagree: “mitotic” clocks tied to cell division can decelerate while “mortality” clocks accelerate in the same person.10
The epigenetic story is the messiest of the three and is where the “accelerated” framing breaks down most clearly.
Why “Advanced” Fits Post-Onset Data Better — and Where That Argument Has Limits
The 2026 review’s central re-reading turns on a distinction popular coverage glosses: the difference between a level-shift and a steeper slope.
Level-shift (advanced aging). Biological-age markers are deviated from controls at first episode. From there, the rate of decline tracks the same as healthy aging — the trajectory lines run parallel, just offset.
Steeper slope (accelerated aging). The gap between patients and controls widens with time. Patients lose ground faster than controls do, year after year.
Longitudinal data favor the level-shift reading for most markers. Brain-age studies find first-episode patients already 1.4–6.9 years “older” than chronological age, with the gap holding steady over 5–10 years of follow-up.2 Cognitive studies show first-episode patients performing like healthy adults 5–10 years older, then tracking mostly stable — except in a subset (around 10% in Thompson 2013) with late decline tied to negative symptoms.2 Telomere shortening shows up early and doesn’t clearly worsen over time.
That reading has a methodological constraint worth naming explicitly. The parallel-trajectory finding is based on measurements starting at first episode. If biological accumulation is occurring during the prodromal period — which typically spans 2–5 years before formal diagnosis — the result would look identical to a static level-shift: a gap present at onset that then tracks parallel afterward.
The review’s own data points toward this possibility. Studies of clinical-high-risk individuals who have not yet received a formal diagnosis already show brain-age deviations of +1.4 to +6.9 years. A static biological shift that appears only at illness onset would not be detectable before onset.
The authors are also careful not to claim the level-shift applies uniformly across all systems. In their own terminology section they note that “true acceleration seems to be less common and may be confined to specific systems” — specifically, rapid brain volume loss in the first 5 years of illness and cumulative oxidative stress accumulation over chronicity.2 The “advanced not accelerated” verdict applies most cleanly to global neuroimaging and cognitive trajectories after onset.
Early Developmental Scars: A Neurodevelopmental Reframe
The review’s most important conceptual contribution is not the terminology debate between “advanced” and “accelerated.” It is the causal model proposed in the discussion section, which the abstract does not capture.
The authors argue that the aging phenotype and the psychiatric diagnosis may both be downstream of the same early developmental insults, rather than schizophrenia directly causing the aging. Three lines of evidence support this framing.
- Widely replicated risk factors for schizophrenia — prenatal stress, childhood adversity, early-life inflammation — are also independent risk factors for telomere shortening, metabolic disease, and accelerated biological aging.2
- Animal models show that perinatal stressors prime offspring for systemic features that closely mirror the schizophrenia aging phenotype: neurostructural abnormalities, glucose-insulin dysregulation, cognitive deficits, and visceral obesity.2
- If early life stress is the common antecedent, abnormal biological aging would be apparent from illness onset — because both conditions reflect the same upstream damage rather than one causing the other, which is exactly the pattern the data show.
The authors describe the multisystem aging in schizophrenia as “the long-term trajectory of early developmental scars” that may also confer increased vulnerability to progressive degeneration, both physical and mental.2
The practical implication is significant. Asking whether schizophrenia causes accelerated aging may be a partially wrongly framed question. Both the psychiatric illness and the biological aging may be expressions of the same upstream developmental exposure. The most upstream intervention — targeting prenatal stress, childhood adversity, early inflammatory insults — would address both the psychiatric and the somatic risk simultaneously, rather than treating aging consequences after illness onset.
What the Antipsychotic Evidence Suggests
The most-asked question is whether antipsychotic medications speed up aging in schizophrenia. Two separate questions often get collapsed here: whether antipsychotics are the origin of the aging phenotype, and whether they worsen or accelerate it once present. The evidence addresses the first; it does not cleanly resolve the second.
Several aging signals are present in antipsychotic-naïve first-episode patients. Drug-naïve samples show impaired glucose regulation, elevated inflammatory cell counts, lower free testosterone, shortened telomere length, and brain-age elevations of +1.4 years or more.2 The biological deviation predates chronic medication. Antipsychotics are not the origin.
That finding does not exonerate them on the second question. A signal existing before an exposure doesn’t mean the exposure doesn’t add to it.
Second-generation agents like olanzapine and clozapine produce weight gain, insulin resistance, and dyslipidemia in a substantial fraction of users. Cumulative anticholinergic exposure from some antipsychotics and adjuncts appears to drive part of the early cognitive decline that fed the original “accelerated aging” framing.2 The review concedes antipsychotics amplify specific subdomains without resolving how large that amplification is over a decade or more of treatment.
Where this lands: stopping antipsychotics is not the answer — they reduce suicide and accident mortality in a way that more than offsets the metabolic cost for most patients. Prevention focuses on metabolic monitoring, lifestyle support, and minimizing anticholinergic burden where clinically possible.
Gaps in the Schizophrenia Aging Literature
The “advanced not accelerated” conclusion has a baseline problem. The parallel-trajectory evidence starts at first episode. Prodromal accumulation spanning the 2–5 years before diagnosis would produce an identical pattern: a gap present at onset that then tracks parallel to controls. The review does not address this directly, and the clinical-high-risk data — showing deviations already present before formal diagnosis — is consistent with pre-onset accumulation rather than a static level-shift that appears only at illness onset.
Genetic confounding is unresolved. Schizophrenia polygenic risk variants overlap with inflammatory, oxidative-stress, and metabolic pathways that independently influence biological aging. Even drug-naïve first-episode data cannot fully disentangle “schizophrenia caused this” from “the shared genetic architecture caused both.” Mendelian randomization designs — which use genetic variants as a natural experiment to test causal direction — can probe this but produce mixed findings across biomarkers.
Survival bias distorts older-cohort data. Adults studied at 60+ are the ones who lived long enough to be studied. If the most vulnerable die earliest, surviving samples look artificially healthy on aging biomarkers, flattening the apparent trajectory.
Antipsychotic confounding is hard to untangle in chronic samples. Most chronic-stage data come from medicated patients; drug-naïve samples are smaller and shorter-followed. “Natural history of schizophrenia aging” is mostly treated schizophrenia aging.
Aging biomarkers were calibrated on healthy people. Brain-age algorithms and epigenetic clocks were trained on cohorts without major psychiatric illness, so applied to schizophrenia they may pick up illness-, treatment-, and lifestyle-related effects. The review explicitly acknowledges these measures “may reflect cumulative biological burden rather than ageing per se.”2
Smoking explains a large share of the signal. Schizophrenia smoking rates are 60–80%. GrimAge and several inflammatory markers largely disappear after smoking adjustment — part of the “aging effect” is a smoking effect.
Epigenetic clocks disagree with each other. Different clocks give different answers in the same patients. Pooled effects across all epigenetic clocks are hard to interpret until the field reconciles what each clock measures.
Smoking Cessation, Metabolic Monitoring, and Upstream Prevention
If the deviation arrives at or before illness onset, the highest-leverage interventions happen at or near first contact with mental-health services rather than years into chronic illness. And if early developmental insults are the deeper causal structure, interventions targeting those exposures — prenatal care, childhood adversity, early inflammatory burden — may address both the psychiatric and somatic risk before either becomes established.
- Metabolic monitoring at first episode. Weight, lipids, glucose, and blood pressure tracked from first contact, not deferred until medication-related weight gain is obvious. Some of the metabolic abnormality is present before any antipsychotic exposure.
- Smoking cessation is the single largest movable risk. Smoking rates in schizophrenia run 60–80%, and several aging signals (GrimAge acceleration, inflammatory markers, mortality risk itself) shrink substantially after smoking adjustment. Tobacco-cessation programs adapted for serious mental illness work; uptake remains low.
- Antipsychotic choice matters over decades. Aripiprazole and ziprasidone produce less weight gain than olanzapine or quetiapine. Where efficacy and tolerability allow, leaning toward lower-metabolic-burden agents compounds into a different cardiovascular trajectory over years.
- Anticholinergic burden is worth tracking. Cumulative exposure to anticholinergic drugs appears to drive part of the early cognitive decline. Reducing exposure where clinically possible is a reasonable prevention move.
- Cardiovascular prevention covers most of the dementia signal. The 2–2.5x dementia risk is partly mediated by cardiovascular disease and anticholinergic burden. Standard prevention (blood pressure, lipids, glucose, exercise) addresses the largest mediated piece.
Common Questions about Schizophrenia and Aging
Does schizophrenia really make you age faster?
It is associated with earlier biological aging, but the mechanism and causal structure are contested. The 2026 systematic review argues “advanced aging” (an early level-shift) fits post-onset trajectory data better than “accelerated aging” (a widening gap). But that verdict only holds from first episode onward — brain-age deviations are measurable in clinical-high-risk individuals before diagnosis. The review’s own authors propose that the aging phenotype may reflect early developmental insults shared with schizophrenia rather than the illness directly causing aging.2
Does schizophrenia cause the aging, or is something else responsible?
The review’s most important nuance is that the question may be partly wrongly framed. The authors propose that widely replicated schizophrenia risk factors — prenatal stress, childhood adversity, early inflammation — are also independent drivers of telomere shortening, metabolic disease, and biological aging. If early developmental insults are a common antecedent for both the psychiatric illness and the aging phenotype, then neither caused the other in a simple sense. Both may reflect the same upstream damage — what the review calls “the long-term trajectory of early developmental scars.”2
How much earlier do people with schizophrenia die?
Roughly 15–20 years on average. Standardized mortality ratios run 2.5–3.5x the general population, with cardiovascular disease as the leading cause, followed by suicide and accidents. The gap holds across countries with very different healthcare systems.6,7
Do antipsychotics cause the aging effect?
Not entirely — and the question has two parts. Several aging markers (telomere shortening, inflammatory abnormalities, glucose dysregulation, brain-age elevation) are present in drug-naïve first-episode patients before chronic antipsychotic exposure, so antipsychotics are not the origin. Whether they worsen the trajectory over time is a separate question the evidence does not cleanly answer. Antipsychotics worsen the metabolic picture specifically, and cumulative anticholinergic burden contributes to cognitive decline. The drugs amplify some subdomains without having created the phenotype.2
How much faster is the brain aging in schizophrenia?
Brain-imaging models estimate brains in schizophrenia look about 3.5 years older than chronological age on average (ENIGMA meta-analysis, 26 cohorts, 95% CI 2.91–4.19). The gap shows up at first episode, ranges from +1.4 to +6.9 years across studies, and tends to hold rather than widen over follow-up — though deviations are already present in pre-diagnosis clinical-high-risk samples.4
Are telomeres shorter in schizophrenia?
Yes — small-to-medium effect across 4 meta-analyses (SMD around −0.39 to −0.52). Shortening shows up in drug-naïve samples, so it is not purely a treatment effect.3,2
Does schizophrenia raise dementia risk?
Yes, by roughly 2.2–2.5x after adjusting for cardiovascular risk factors, with dementia arriving roughly 20 years earlier on average. Cumulative anticholinergic exposure is now thought to account for a meaningful share of the early cognitive decline.2
Can the aging effect be slowed?
The metabolic and cardiovascular components are modifiable: smoking cessation, blood-pressure and lipid control, exercise, weight management, lower-metabolic-risk antipsychotics where clinically appropriate. The earlier these start, the more the level-shift can be partly offset. If the review’s neurodevelopmental framing is right, the deeper leverage point is earlier still — prenatal and early-childhood exposures that set the trajectory for both the illness and the aging.
Why do brain age and epigenetic clocks sometimes disagree?
They measure different things. Brain age tracks structural MRI features; different epigenetic clocks track different biological signals — some intrinsic cellular aging, others mortality risk, others cumulative damage. In schizophrenia some clocks show acceleration while others show youth-stage deceleration. That mixed picture is part of why “advanced” fits better than a uniform “accelerated” verdict.9,10
References
- Is schizophrenia a syndrome of accelerated aging? Kirkpatrick B et al. Schizophrenia Bulletin. 2008;34(6):1024–1032. doi:10.1093/schbul/sbm140
- Schizophrenia and accelerated ageing: systematic review and future research directions. Fernandez-Egea E et al. British Journal of Psychiatry. 2026:1–13. doi:10.1192/bjp.2026.10606
- Leukocyte telomere length in patients with schizophrenia and related disorders: a meta-analysis of case-control studies. Ayora M et al. Molecular Psychiatry. 2022;27(6):2968–2975. doi:10.1038/s41380-022-01541-7
- Brain ageing in schizophrenia: evidence from 26 international cohorts via the ENIGMA Schizophrenia consortium. Constantinides C et al. Molecular Psychiatry. 2023;28(3):1201–1209. doi:10.1038/s41380-022-01897-w
- Brain age in mood and psychotic disorders: a systematic review and meta-analysis. Ballester PL et al. Acta Psychiatrica Scandinavica. 2022;145(1):42–55. doi:10.1111/acps.13371
- Premature mortality in adult schizophrenia. Olfson M et al. JAMA Psychiatry. 2015;72(12):1172–1181. doi:10.1001/jamapsychiatry.2015.1737
- A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Saha S et al. Archives of General Psychiatry. 2007;64(10):1123–1131. doi:10.1001/archpsyc.64.10.1123
- Schizophrenia: a systemic disorder. Kirkpatrick B et al. Clinical Schizophrenia & Related Psychoses. 2014;8(2):73–79. doi:10.3371/CSRP.KIMI.031513
- A systematic review and meta-analysis of epigenetic clocks in schizophrenia. Chrusciel JH et al. Schizophrenia Research. 2022;246:172–174. doi:10.1016/j.schres.2022.06.029
- A computational solution for bolstering reliability of epigenetic clocks: implications for clinical studies. Higgins-Chen AT et al. Nature Aging. 2022;2:644–661. doi:10.1038/s43587-022-00248-2