
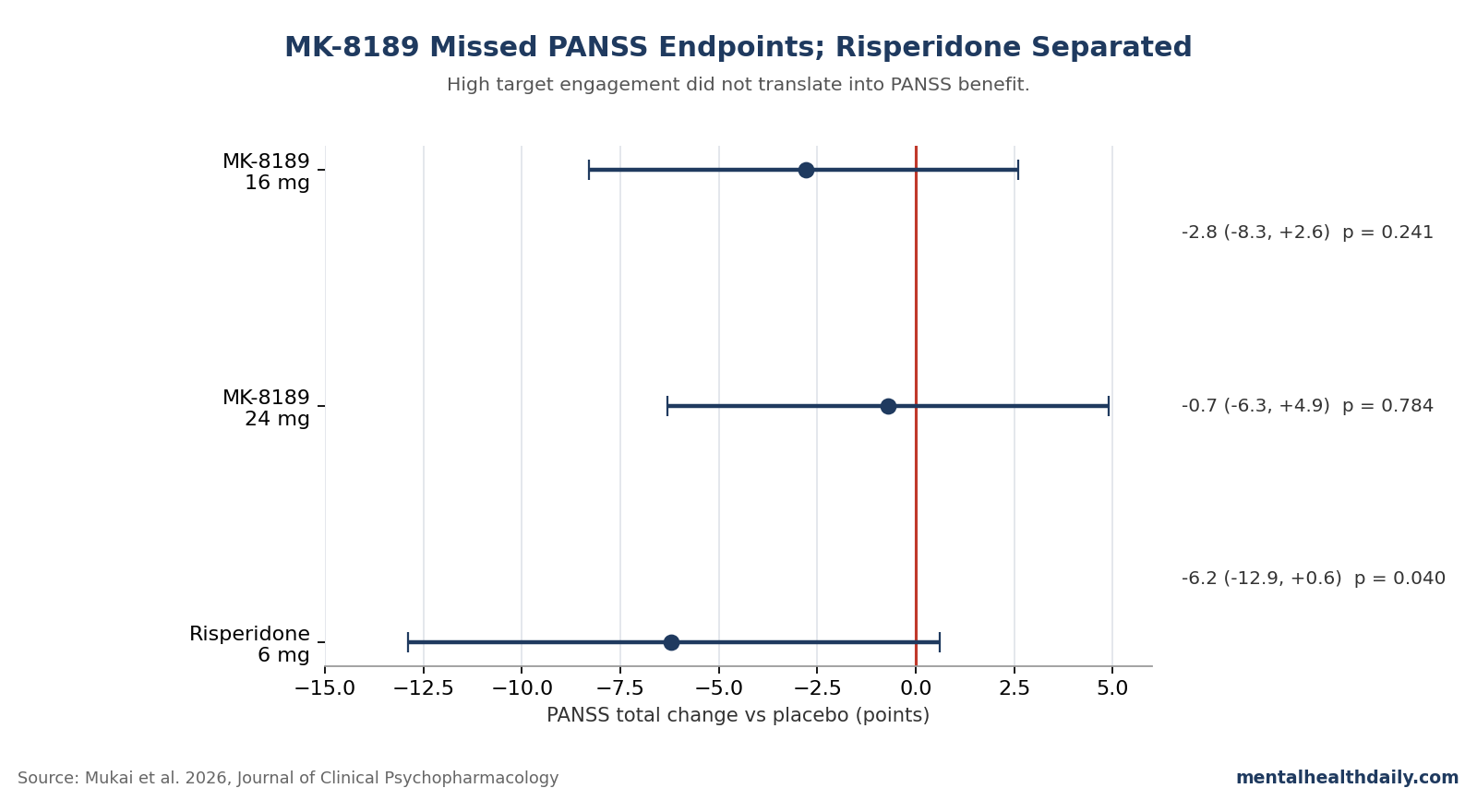
Mukai et al.’s 499-person Phase 2b schizophrenia trial tested whether higher, sustained PDE10A inhibition could turn MK-8189 into a non-D2 antipsychotic; it did not. At week 6, MK-8189 missed the PANSS total endpoint at 16 mg (−2.8 points vs. placebo; 95% CI −8.3 to 2.6; P = 0.241) and 24 mg (−0.7 points; 95% CI −6.3 to 4.9; P = 0.784), while risperidone separated by −6.2 points (P = 0.040).1
Research Highlights
- MK-8189 failed at both tested doses. The prespecified 6-week PANSS total comparisons were −2.8 points for 16 mg and −0.7 points for 24 mg vs. placebo, with both 95% CIs crossing 0.1
- The trial could detect an antipsychotic signal. Risperidone 6 mg, included for assay sensitivity, beat placebo by −6.2 PANSS points (95% CI −12.9 to 0.6; P = 0.040).1
- Higher target higher engagement failed to rescue the mechanism. MK-8189 24 mg was predicted to produce around 80% sustained PDE10A occupancy, the highest continuous 24-hour engagement tested for this target, yet its symptom effect was numerically weaker than 16 mg.1
- The tolerability pattern moved opposite the premise. Acute-period extrapyramidal symptoms reached 22.7% on 24 mg and 15.2% on 16 mg, vs. 3.9% on placebo and 6.2% on risperidone.1
- Weight separated, but psychosis did not. By week 12, MK-8189 groups lost 4.3 to 5.4 kg while risperidone gained 3.0 kg, a real metabolic signal that still does not make MK-8189 an antipsychotic.1
PDE10A is phosphodiesterase 10A, an enzyme concentrated in striatal medium spiny neurons, the cells that route movement, reward, and psychosis-relevant dopamine signaling through direct and indirect output pathways. The drug-development bet was that inhibiting PDE10A could imitate useful parts of dopamine D2 blockade downstream, inside striatal signaling circuits, without directly blocking D2 receptors across the brain.1
The 2026 trial pushes that idea close to a stopping point for MK-8189-like monotherapy. Contrary to the mechanism prediction, more sustained enzyme higher engagement failed to produce more antipsychotic efficacy; it produced more discontinuation and more motor adverse events.
Why MK-8189 Was a Serious Test of PDE10A
Dopamine D2 receptor blockade has anchored schizophrenia pharmacology for decades. Standard antipsychotics vary in serotonin, histamine, muscarinic, and adrenergic effects, but clinical antipsychotic efficacy still usually tracks D2 receptor engagement.10
PDE10A inhibition was attractive because it sat downstream from dopamine receptors. PDE10A breaks down the second messengers cyclic adenosine monophosphate and cyclic guanosine monophosphate, often shortened to cAMP and cGMP.
Those molecules carry intracellular signals after receptors at the cell surface have been activated.
Striatal signaling target: in striatal medium spiny neurons, changing cAMP/cGMP signaling could theoretically activate the same indirect pathway that D2 antagonists influence, while also engaging direct-pathway neurons that might help cognition or negative symptoms.
That was the clean pharmacology pitch: treat positive symptoms, maybe help broader schizophrenia domains, and avoid pituitary, cortical, metabolic, and motor costs from direct D2 blockade.4,5
MK-8189 was not a casual probe of that hypothesis. Merck developed it as a controlled-release PDE10A inhibitor to flatten peak-to-trough exposure and sustain target engagement.
An earlier 12 mg proof-of-concept trial produced a trend toward PANSS total improvement vs. placebo at 4 weeks (−4.7 points; 95% CI −9.8 to 0.5; P = 0.074), enough to justify a larger, longer, higher-dose test.6
The Phase 2b Trial Put the Dose-Engagement Argument on Trial
The follow-up trial enrolled adults ages 18 to 55 with active-phase schizophrenia across 75 centers in 11 countries from December 2020 to June 2024. Participants needed a PANSS total score of at least 80 and a Clinical Global Impression-Severity score of at least 4, meaning at least moderately ill.
Mean baseline CGI-S was 5.1, corresponding to “markedly ill.”1
The acute phase randomized participants to MK-8189 8 mg, 16 mg, 24 mg, risperidone 6 mg, or placebo. The 8 mg arm was dropped after 41 participants and excluded from model-based efficacy analyses, leaving 132 participants treated with 16 mg, 132 with 24 mg, 65 with risperidone, and 129 with placebo in the main acute-period arms.1
PANSS means Positive and Negative Syndrome Scale, a 30-item clinician-rated symptom scale for schizophrenia. Lower scores after treatment mean improvement.
The trial’s primary outcome was PANSS total change from baseline at 6 weeks.
The power assumptions were favorable to MK-8189: researchers expected a placebo-adjusted PANSS total difference of −6.56 points for 16 mg and −8.44 points for 24 mg. With 128 planned participants per main arm and a 35% dropout assumption, the trial had a 91% probability of declaring success for at least 1 MK-8189 dose if those effects were real.1
The observed results went the other way:
- MK-8189 16 mg vs. placebo: −2.8 PANSS total points (95% CI −8.3 to 2.6; P = 0.241).
- MK-8189 24 mg vs. placebo: −0.7 points (95% CI −6.3 to 4.9; P = 0.784).
- Risperidone 6 mg vs. placebo: −6.2 points (95% CI −12.9 to 0.6; P = 0.040).
Risperidone separated. MK-8189 did not.
That active-control result reduces the usual escape hatch for a failed antipsychotic trial: the study was not too noisy to detect any drug-placebo difference.
Secondary Symptom Measures Did Not Rescue the Trial
PANSS positive symptoms, the subscale most directly tied to delusions, hallucinations, suspiciousness, conceptual disorganization, and related psychosis symptoms, also failed to show a confirmed MK-8189 advantage. At week 6, the 16 mg arm differed from placebo by −1.3 points (95% CI −3.1 to 0.5), while the 24 mg arm differed by −1.4 points (95% CI −3.2 to 0.5).1
CGI-S, the clinician’s global severity rating, was similarly flat. MK-8189 16 mg differed from placebo by −0.2 points (95% CI −0.5 to 0.1), and MK-8189 24 mg differed by 0.0 points (95% CI −0.3 to 0.3).
The researchers also reported that subgroup analyses by region, race, and sex all had 95% CIs including 0 for MK-8189 vs. placebo.1
The important detail is more than non-significance. The 24 mg dose, which should have been the stronger mechanistic test, was not directionally better than 16 mg on PANSS total, PANSS positive symptoms, or CGI-S.
For a target-engagement rescue story, that is the wrong dose-response shape.
Motor Side Effects Undercut the Dopamine-Sparing Claim
The tolerability signal is the part of the trial that makes the negative efficacy harder to dismiss. MK-8189 was supposed to avoid direct D2 blockade and therefore avoid much of the motor-liability profile associated with conventional antipsychotic pharmacology.
The acute-period adverse-event table did not look dopamine-sparing.
Extrapyramidal symptoms, or EPS, are drug-induced movement problems such as stiffness, tremor, akathisia, and dystonia. In the 6-week acute period, EPS composites were reported in 22.7% of participants on MK-8189 24 mg and 15.2% on 16 mg, compared with 3.9% on placebo and 6.2% on risperidone.1
Dystonia, a sustained or spasmodic muscle-contraction reaction, showed the same dose-related pattern: 12.1% on MK-8189 24 mg, 5.3% on 16 mg, 0.8% on placebo, and 0.0% on risperidone. Akathisia reached 9.1% on MK-8189 24 mg and 6.1% on 16 mg, vs. 1.6% on placebo and 6.2% on risperidone.1
Discontinuation due to adverse events also moved against the higher dose. During the acute period, 25.0% of participants on MK-8189 24 mg discontinued for an adverse event, compared with 12.9% on 16 mg, 12.3% on risperidone, and 12.4% on placebo.
The researchers wrote that the hoped-for improved tolerability profile vs. current antipsychotics “was not supported by the findings.”1
Weight Loss Was Real but Not Enough
MK-8189 did separate from risperidone on weight. At week 12, participants on MK-8189 16 mg lost 5.4 kg on average and those on 24 mg lost 4.3 kg, while the risperidone group gained 3.0 kg.
The MK-8189 vs. risperidone differences were −8.5 kg and −7.3 kg, both nominally significant at P < 0.001.1
That finding is biologically interesting because PDE10A signaling has also been linked to adipocyte and metabolic biology.9 It also replicates the earlier MK-8189 observation that weight moved down while risperidone moved up.6
But a favorable weight trajectory does not compensate for absent antipsychotic efficacy in an acute-schizophrenia monotherapy trial. The practical drug-development question was whether MK-8189 could treat psychosis while improving the metabolic tradeoff.
The trial answered only the second half in the desired direction.
Prior PDE10A Trials Make the Failure Look Mechanism-Level
MK-8189 is now the third PDE10A monotherapy program to fail a confirmed acute-schizophrenia efficacy test. Walling et al. tested PF-02545920, also known as balipodect, and concluded that PDE10A inhibitor monotherapy was not an effective treatment for acute schizophrenia.2
Macek et al. tested TAK-063 and found only a small, nonsignificant PANSS total effect vs. placebo.3
Before the Mukai 2026 result, those failures could still be explained away. PF-02545920 and TAK-063 likely produced only modest sustained 24-hour PDE10A inhibition.
The 2024 MK-8189 proof-of-concept signal at 12 mg kept alive the possibility that the target required higher, steadier occupancy.6
The 24 mg arm tested that rescue hypothesis directly. Mukai et al. estimated that it produced around 80% sustained enzyme occupancy, which they described as the highest continuous 24-hour target engagement evaluated for a PDE10A inhibitor.
The researchers concluded that the lack of benefit “strongly suggests that PDE10A inhibition, by itself, is not a useful approach for treating acute psychosis in people with schizophrenia.”1
That wording is calibrated. It does not rule out every possible PDE10A-adjacent idea: add-on therapy, cognition-focused trials, negative-symptom enrichment, or different kinetic profiles remain different questions.
It does make MK-8189-like PDE10A monotherapy for acute psychosis look exhausted.
KarXT Shows the Non-D2 Door Is Still Open
Non-D2 contrast: the MK-8189 result should not be flattened into “non-D2 antipsychotics failed.” Xanomeline-trospium, the muscarinic M1/M4 agonist-antagonist combination often called KarXT and marketed as Cobenfy, separated from placebo in Phase 3 schizophrenia trials and became the first approved antipsychotic with a mechanism not built around D2 blockade.7,8
KarXT’s contrast with MK-8189 is useful because both programs asked whether psychosis could be treated without directly blocking dopamine D2 receptors. Muscarinic M1/M4 signaling appears capable of shifting cholinergic-dopaminergic balance enough to produce clinical antipsychotic efficacy.
PDE10A inhibition, even at high sustained engagement, did not.
Other non-D2 approaches remain uneven. Ulotaront, a trace amine-associated receptor 1 agonist, produced positive Phase 2 data in schizophrenia, but later development has been more difficult.11
Glycine-site and other glutamatergic strategies have also produced repeated disappointments. The sober 2026 read is selective optimism: the post-D2 era exists, but receptor logic that looks elegant in animals still has to survive human symptom endpoints.
Three Caveats Keep the Result Narrow
- Placebo response was high. Placebo PANSS total improved by 17.8 points at week 6. Mukai et al. noted that the prior Phase 2a placebo change was around 10 points at week 4, and COVID-era schizophrenia trials have shown elevated placebo response. High placebo response can compress drug-placebo separation, although risperidone still separated in this trial.1
- Cognition and negative symptoms were not the main test. The trial used PANSS total in acute psychosis as the primary endpoint. PDE10A’s preclinical case included cognition and negative-symptom hypotheses, but the study did not use a cognition-enriched or negative-symptom-enriched design.1
- Combination therapy was not tested. Preclinical work has explored PDE10A inhibition with risperidone-like signaling, but this trial tested MK-8189 as monotherapy against placebo and an active control. A lower-dose D2 add-on concept remains scientifically possible, even if the commercial case is much weaker after this result.5
Questions About MK-8189, PDE10A, and Schizophrenia Trials
Did MK-8189 fail because the trial was insensitive?
That is unlikely. Risperidone 6 mg beat placebo by −6.2 PANSS points at week 6 (P = 0.040), which means the trial could detect a known antipsychotic effect.
MK-8189’s failure happened inside an assay-sensitive study, not a study where every active drug looked inert.1
Did the higher 24 mg dose look better than 16 mg?
No. The 24 mg dose was predicted to produce around 80% sustained PDE10A occupancy, but its week-6 PANSS total difference vs. placebo was only −0.7 points, compared with −2.8 points for 16 mg.
Higher engagement brought more EPS and discontinuation, not more efficacy.1
Were any symptom subgroups clearly positive for MK-8189?
No clear subgroup rescue appears in the paper. The researchers reported that subgroup analyses by region, race, and sex all had 95% CIs including 0 for MK-8189 vs. placebo, and key secondary symptom endpoints also failed to confirm benefit.1
Why did MK-8189 reduce weight if it failed on psychosis?
PDE10A biology may affect metabolic signaling separately from antipsychotic efficacy. In this trial, weight moved favorably: −5.4 kg on 16 mg and −4.3 kg on 24 mg at week 12, while risperidone gained 3.0 kg.
That does not prove symptom efficacy, but it explains why the metabolic result should not be dismissed as noise.1,9
Does this result rule out all PDE10A use in schizophrenia?
It rules out the strongest current case for MK-8189-like PDE10A monotherapy in acute psychosis. It does not directly test add-on use, cognition-focused endpoints, or negative-symptom-enriched trials.
Those would need different designs and a reason to accept the motor-side-effect signal seen at higher MK-8189 exposure.
Is MK-8189 related to Cobenfy or KarXT?
No. MK-8189 is a PDE10A inhibitor.
Cobenfy is xanomeline-trospium, a muscarinic M1/M4 receptor agonist strategy paired with peripheral muscarinic blockade to reduce side effects. Both are non-D2 approaches to schizophrenia, but their mechanisms and trial outcomes are different.7,8
References
- Mukai Y, Kost J, Snow-Adami L, Reed J, Taylor T, Kent J, Egan MF. Phase 2b trial of the PDE10A inhibitor MK-8189 in people with an acute episode of schizophrenia. J Clin Psychopharmacol. 2026;46(3):311–319. doi:10.1097/jcp.0000000000002152
- Walling DP, Banerjee A, Dawra V, et al. Phosphodiesterase 10A inhibitor monotherapy is not an effective treatment of acute schizophrenia. J Clin Psychopharmacol. 2019;39(6):575–582. doi:10.1097/jcp.0000000000001128
- Macek TA, McCue M, Dong X, et al. A phase 2, randomized, placebo-controlled study of the efficacy and safety of TAK-063 in subjects with an acute exacerbation of schizophrenia. Schizophr Res. 2019;204:289–294. doi:10.1016/j.schres.2018.08.028
- Menniti FS, Chappie TA, Humphrey JM, et al. Phosphodiesterase 10A inhibitors: a novel approach to the treatment of the symptoms of schizophrenia. Curr Opin Investig Drugs. 2007;8(1):54–59. PubMed
- Grauer SM, Pulito VL, Navarra RL, et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331(2):574–590. doi:10.1124/jpet.109.155994
- Mukai Y, Lupinacci R, Marder S, et al. Effects of PDE10A inhibitor MK-8189 in people with an acute episode of schizophrenia: a randomized proof-of-concept clinical trial. Schizophr Res. 2024;270:37–43. doi:10.1016/j.schres.2024.05.019
- Brannan SK, Sawchak S, Miller AC, Lieberman JA, Paul SM, Breier A. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. N Engl J Med. 2021;384(8):717–726. doi:10.1056/nejmoa2017015
- Kaul I, Sawchak S, Walling DP, et al. Efficacy and safety of xanomeline-trospium chloride in schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2024;81(8):749–756. doi:10.1001/jamapsychiatry.2024.0785
- Hankir MK, Kranz M, Gnad T, et al. A novel thermoregulatory role for PDE10A in mouse and human adipocytes. EMBO Mol Med. 2016;8(7):796–812. doi:10.15252/emmm.201506085
- Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951–962. doi:10.1016/s0140-6736(13)60733-3
- Koblan KS, Kent J, Hopkins SC, et al. A non-D2-receptor-binding drug for the treatment of schizophrenia. N Engl J Med. 2020;382(16):1497–1506. doi:10.1056/nejmoa1911772