
A 2026 mechanistic Alzheimer’s study found that suppressing NDST3 shifted lysosomal pH in APP695Swe neuronal cells from 5.6 back below 5.0, then reduced amyloid-β, MAPT/tau pathology, neuronal injury, and memory deficits in 3xTg-AD mice.1
Research Highlights
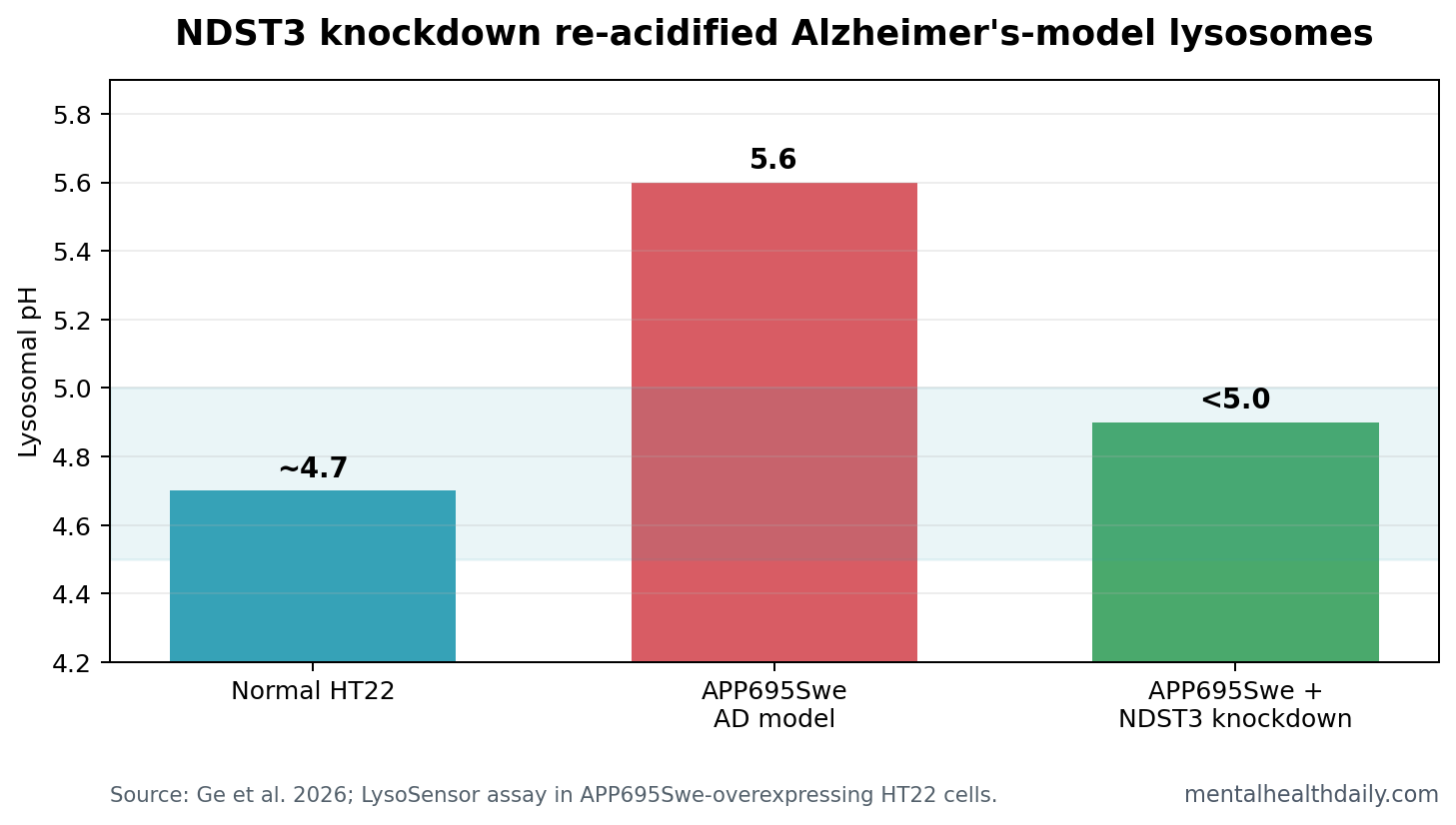
- Lysosome pH moved back down: APP695Swe-overexpressing HT22 cells had lysosomal pH around 5.6, while NDST3 knockdown re-acidified lysosomes to below 5.0.
- Human AD tissue showed the same direction: postmortem brain staining found higher NDST3 in 3 Alzheimer’s disease samples than in 3 control samples (p = 0.0022).
- Autophagic clearance improved: NDST3 knockdown increased V-ATPase assembly, raised cathepsin B activity, improved dextran degradation, and reduced amyloid-β and MAPT/tau readouts.
- Mouse pathology fell: 3xTg-Ndst3+/- mice had fewer poorly acidified autolysosomes, lower APP-CTFβ and MAPT/tau burden, and reduced plaque/tau staining vs. 3xTg-AD mice.
- Translation remains early: this is cell, mouse, and small postmortem-tissue evidence; it does not show that NDST3 inhibition treats human Alzheimer’s disease.
Lysosomes are acidic recycling compartments that help neurons degrade damaged proteins and other cellular cargo. Autophagy is the cleanup pathway that sends this cargo to lysosomes. In Alzheimer’s disease, that system matters because amyloid-β and MAPT/tau are plaque and tangle markers as well as proteins that neurons must process, clear, or fail to clear.
NDST3 is a tubulin deacetylase, meaning it can remove acetyl groups from α-tubulin, a structural protein in microtubules. Ge et al. tested a specific chain: NDST3 affects perinuclear microtubule acetylation, microtubule acetylation affects V-ATPase assembly on lysosomes, V-ATPase assembly affects lysosomal acidity, and acidity affects whether Alzheimer’s-related proteins are degraded efficiently.
NDST3 Knockdown Shifted Lysosomal pH From 5.6 to Below 5.0
Lysosomal acidification means keeping the lysosome’s interior acidic enough for digestive enzymes to work. The 2026 study used LysoSensor Yellow/Blue DND-160, a fluorescent pH probe, to measure lysosomal pH in mouse hippocampal HT22 cells.
Normal HT22 cells had lysosomal pH around 4.7. APP695Swe-overexpressing cells, a neuronal Alzheimer’s model built around mutant amyloid precursor protein, shifted upward to pH 5.6. That is less acidic, and less acidic lysosomes are worse places for protein degradation.
Silencing Ndst3 reversed much of that alkalinization: the APP695Swe cells treated with shNdst3 fell back below pH 5.0. The study also found more lysosome-bound V-ATPase V1 subunits after NDST3 knockdown, supporting the proposed mechanism rather than only a descriptive pH change.
V-ATPase Assembly Is the Mechanistic Link
V-ATPase is the proton pump that acidifies lysosomes by moving H+ into the lysosomal lumen. It works as a V1-V0 holoenzyme: V0 sits in the membrane, while V1 docks to help drive proton pumping. If fewer V1 subunits assemble on the lysosome, acidification can fail.
Ge et al. isolated lysosomal fractions and measured V1 subunits ATP6V1A and ATP6V1C1 relative to the V0 subunit ATP6V0D. APP695Swe cells had lower lysosomal V1 docking than normal cells. NDST3 knockdown reversed that decrease, while total whole-cell levels of the V-ATPase proteins stayed unchanged.
Mechanism handle: the intervention did not appear to make cells produce more V-ATPase everywhere. It changed assembly at lysosomes, which fits the microtubule-acetylation model.
That detail is important because broad lysosomal acidification is not automatically good. The earlier NDST3 work showed that losing NDST3 can over-acidify lysosomes and worsen C9orf72-related proteotoxic stress in ALS/FTD models.5 In the Alzheimer’s setting, the problem was different: lysosomes were too alkaline, so partial NDST3 suppression moved pH toward the working range.
Amyloid and Tau Burden Fell After Lysosomal Function Improved
Better lysosomal acidity only matters for Alzheimer’s biology if it changes the protein-clearance problem. Ge et al. tested that with several linked readouts.
Cathepsin B activity: cathepsin B is a lysosomal protease, an enzyme that helps degrade proteins inside lysosomes. APP695Swe cells had weaker Magic Red cathepsin B signal, while shNdst3 treatment increased the signal without simply increasing LAMP2-marked lysosome abundance or cathepsin B colocalization.
Dextran degradation: the researchers used AF647-dextran to test lysosomal degradative capacity. APP695Swe cells retained more fluorescent dextran after a 4-hour chase, indicating poorer degradation. NDST3 knockdown improved the degradation pattern.
Autophagic flux: APP695Swe cells showed a disrupted LC3B turnover pattern and accumulated p62, both consistent with late-stage autophagy-lysosome impairment. NDST3 knockdown reduced the LC3B-II/LC3B-I ratio and restored chloroquine-sensitive LC3B-II accumulation, which points toward improved flux rather than simple autophagy activation.
Protein-clearance readouts: extracellular amyloid-β40 and amyloid-β42 were elevated in APP695Swe cells and decreased after shNdst3 treatment. APP-FL and APP-CTFβ, direct precursors in the amyloid pathway, also fell after NDST3 knockdown, and chloroquine blocked that reduction.
Tau readouts followed the same logic: total MAPT/tau and phosphorylated MAPT/tau were reduced after shNdst3 in MAPT/tau P301L cells, and chloroquine again abolished the effect.
Human Tissue Supported Relevance, But Not Treatment Efficacy
Ge et al. looked beyond cell models by staining NDST3 in mouse and human brain tissue. NDST3 protein was elevated in hippocampal CA1, CA2, CA3, and dentate gyrus regions of 3xTg-AD mice at 3, 6, and 12 months vs. nontransgenic controls.
Postmortem human brain samples pointed in the same direction. NDST3 staining was higher in Alzheimer’s disease tissue than control tissue, with 3 human subjects per group and p = 0.0022. That supports biological relevance, but the sample is too small to estimate diagnostic value, disease-stage specificity, or treatment response.
Evidence-strength note: human tissue staining can show that a target is altered in disease tissue. It cannot show whether changing that target in living patients would slow Alzheimer’s disease, improve cognition, or avoid toxicity.
Partial Ndst3 Knockout Improved Mouse Pathology and Memory
The strongest disease-model evidence came from 3xTg-AD mice crossed with Ndst3 knockout mice. The resulting 3xTg-Ndst3+/- mice had lower hippocampal NDST3, no obvious general-health or survival problems during the study period, and no major serum-biochemistry toxicity signal in the reported assays.
At 10 months, 3xTg-AD mice had more poorly acidified autolysosomes in the hippocampus than nontransgenic controls. 3xTg-Ndst3+/- mice showed fewer poorly acidified autolysosomes, lower p62 and LC3B-II/I readouts, reduced APP-CTFβ and MAPT/tau, fewer amyloid-β plaques, and less p-MAPT/tau Ser422 staining.
Neuronal structure also improved. Compared with 3xTg-AD controls, 3xTg-Ndst3+/- mice had less Fluoro-Jade C/NeuN overlap, denser dendritic spines, better-preserved synaptic ultrastructure, lower microglial activation, and restored GFAP staining in the hippocampus.
Morris water maze: 3xTg-Ndst3+/- mice had shorter escape latencies and swimming distances than 3xTg-AD mice, then spent more time in the target quadrant and crossed the platform location more often during the probe test. Swimming speed was similar across groups, reducing the chance that motor ability explained the result.
Novel object recognition: 3xTg-Ndst3+/- mice reached a discrimination index around 0.4 and explored the novel object more than the familiar one, unlike 3xTg-AD controls.
Other Lysosomal pH Studies Also Reduced Alzheimer’s Pathology
The result does not stand alone. Lee et al. previously reported that faulty autolysosome acidification in Alzheimer’s mouse models induced intraneuronal amyloid-β buildup and contributed to plaque formation.2 Tong et al. tested a different lysosomal ion pathway, TPCN2 inhibition, and reported reduced amyloid pathology plus mitigated memory impairment in Alzheimer’s models.3
Long et al. worked closer to the same microtubule/V-ATPase lane: HDAC6-mediated V-ATPase assembly and lysosomal acidification improved autophagic amyloid-β clearance in Alzheimer’s models.4 Tang et al. supplied the NDST3-specific foundation by showing that NDST3 deacetylates α-tubulin and suppresses V-ATPase assembly and lysosomal acidification.5
Together, these papers sharpen the Alzheimer’s target class. The shared claim is more specific than “increase autophagy”: neuronal lysosomes may fail because pH and ion handling are wrong, and correcting that chemistry can improve protein degradation in preclinical models.
Why NDST3 Is Not a Human Alzheimer’s Treatment Yet
NDST3 looks interesting because it acts near the machinery that acidifies lysosomes and because the 2026 study linked the mechanism to amyloid, tau, neuronal structure, inflammation, and behavior. That breadth makes the target stronger than a single isolated biomarker.
The translation gap is still large:
- No NDST3 drug was tested: partial genetic knockout is not the same as a brain-penetrant, dose-controlled, reversible inhibitor.
- Human evidence was small: 3 AD samples and 3 controls can support direction, not clinical reliability.
- Mouse rescue is not patient rescue: 3xTg-AD mice model parts of amyloid and tau biology, but they do not reproduce the full human disease.
- pH biology can cut both ways: too little acidification impairs degradation, but too much acidification or mistimed acidification can create separate stress.
That last caveat is the most important. NDST3 may be a target only when lysosomes are pathologically under-acidified. A future therapy would need biomarkers showing which cells, disease stages, and patients are in that state.
Questions About NDST3 and Alzheimer’s Disease
Why did NDST3 matter in this Alzheimer’s study?
NDST3 is a tubulin deacetylase that can regulate microtubule acetylation and V-ATPase assembly, which in turn affects lysosomal pH.
Did the study test an Alzheimer’s drug?
No. It tested NDST3 knockdown in cell models and partial Ndst3 knockout in Alzheimer’s-model mice. That is target biology, not a clinical treatment.
Why does lysosomal pH matter for amyloid and tau?
Lysosomal enzymes work best in an acidic compartment. When lysosomes become too alkaline, neurons can lose the ability to degrade amyloid-β precursors and abnormal tau efficiently.
Could NDST3 inhibition be risky?
Yes. Lysosomal pH is basic cell biology. A useful therapy would need to correct disease-related under-acidification without pushing lysosomes into harmful over-acidification or disrupting other tissues.
References
- Ge C, Wang K, Tang H, et al. NDST3 suppression restores lysosomal acidification and ameliorates amyloid-beta and MAPT/tau pathology in Alzheimer’s disease. Translational Neurodegeneration. 2026;15:16. https://doi.org/10.1186/s40035-026-00549-1
- Lee J-H, Yang D-S, Goulbourne CN, et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nature Neuroscience. 2022;25:688-701. https://doi.org/10.1038/s41593-022-01084-8
- Tong BC-K, Wu AJ, Huang AS, et al. Lysosomal TPCN (two pore segment channel) inhibition ameliorates beta-amyloid pathology and mitigates memory impairment in Alzheimer disease. Autophagy. 2022;18:624-642. https://doi.org/10.1080/15548627.2021.1945220
- Long Z, Ge C, Zhao Y, et al. Enhanced autophagic clearance of amyloid-beta via HDAC6-mediated V-ATPase assembly and lysosomal acidification protects against Alzheimer’s disease in vitro and in vivo. Neural Regeneration Research. 2025;20:2633-2644. https://pubmed.ncbi.nlm.nih.gov/38993141/
- Tang Q, Liu M, Liu Y, Hwang R, Zhang T, Wang J. NDST3 deacetylates alpha-tubulin and suppresses V-ATPase assembly and lysosomal acidification. EMBO Journal. 2021;40:e107204. https://doi.org/10.15252/embj.2020107204
- van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. New England Journal of Medicine. 2023;388:9-21. https://doi.org/10.1056/NEJMoa2212948