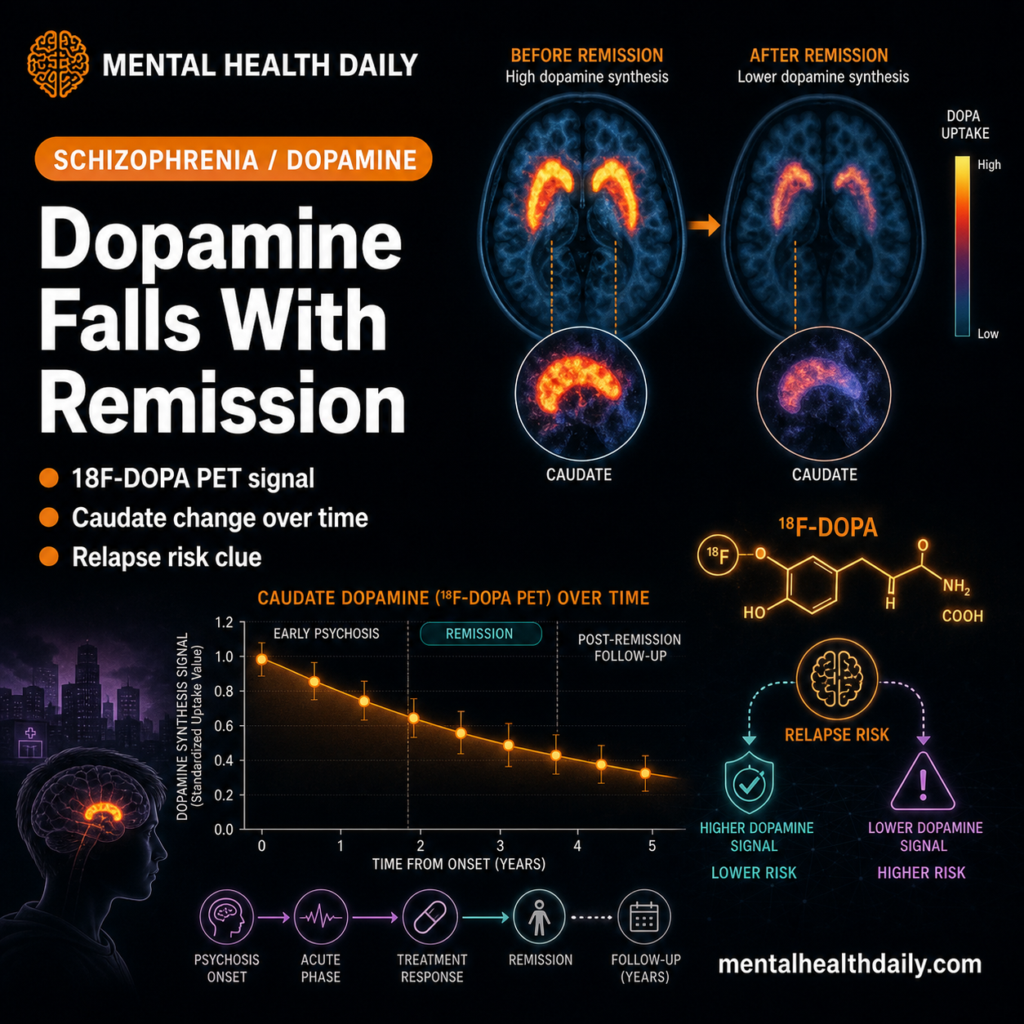
The dopamine hypothesis of schizophrenia is the field’s longest-running mechanistic story, and most of its supporting evidence has been cross-sectional. A 2026 longitudinal PET study by Schulz and colleagues followed the same 28 patients across psychosis and early remission.1
Research Highlights
- The dopamine hypothesis of schizophrenia is the field’s longest-running mechanistic story. Cross-sectional 18F-DOPA PET meta-analyses have shown elevated striatal dopamine synthesis during acute psychosis (typical Cohen’s d ~0.79).2 Whether dopamine synthesis tracks the relapsing-remitting course of psychotic symptoms has been hard to test directly.
- The Schulz 2026 longitudinal PET study followed 28 schizophrenia patients across two scans — one during a psychotic episode, one during early psychotic remission (6 weeks to 12 months later) — alongside 21 matched controls scanned twice.1
- Caudate dopamine synthesis decreased significantly from psychosis to remission in patients (post hoc t = 1.8, p = 0.043), with a significant group-by-session interaction (p = 0.034). The same direction held in nucleus accumbens but didn’t reach significance in post hoc tests.1
- During remission, patients had LOWER striatal dopamine synthesis than controls (caudate t = 2.1, p = 0.02; accumbens t = 2.4, p = 0.01) — not normalization, but undershoot.1
- Higher caudate dopamine synthesis during psychosis predicted relapse within 12 months (F = 4.4, p = 0.049). Of the 22 patients followed up, 32% relapsed, consistent with the standard 25–30% one-year relapse rate even on continued antipsychotics.1
The hypothesis has narrowed substantially over the last 15 years. Early versions framed schizophrenia as a fundamental excess of dopamine. Modern versions, anchored on Howes and Kapur’s 2009 reformulation, locate the disturbance specifically in presynaptic dopamine synthesis and storage in the dorsal (associative) striatum, with normal or reduced activity in mesolimbic and mesocortical pathways.3
Most of the supporting PET evidence has compared different patients to different controls at a single timepoint, usually during active illness. Schulz instead followed the same 28 patients across psychosis and early remission — and produced findings that complicate the simple “more dopamine equals more psychosis” framing.1
Schulz 2026: A Longitudinal 18F-DOPA PET Design
The trigger paper enrolled 28 patients meeting DSM-5 criteria for schizophrenia and 21 matched healthy controls at the Technical University of Munich. All patients had at least one prior psychotic episode (excluding first-episode cases to ensure diagnostic specificity). The core design:1
- Session 1: during acute psychosis. PANSS positive items P1, P3, or P6 had to score ≥ 4 (moderate severity). Patients had been in an acute episode for at least 2 weeks.
- Session 2: during early psychotic remission. Andreasen criteria for remission applied (P1, P2, P3, P6, G5, G9 all ≤ 3) for at least 6 weeks; second scan within 6 weeks to 12 months of session 1.
- Controls scanned at matched intervals. 21 controls had two scans separated by comparable durations.
- Exploratory follow-up at 12 months after session 2 to assess subsequent psychotic relapse.
The PET measure was the influx constant kicer, computed via Gjedde-Patlak graphical analysis with cerebellum as reference region — a standard proxy for striatal dopamine synthesis and storage (DSS). Three subregions were extracted: nucleus accumbens, caudate, and putamen.
Antipsychotic medication was tracked via plasma levels and chlorpromazine equivalents. Patients had not changed antipsychotic regimens between scans in any pre-specified way (mean CPZ equivalent ~470 mg/d during psychosis, ~441 mg/d during remission, not significantly different). Crucially, the main results survived adjustment for CPZ equivalents, plasma levels, smoking status, and individual antipsychotic class — clozapine, aripiprazole, amisulpride.1
Caudate Dopamine Synthesis Drops Across the Course
The headline statistical finding was a significant group-by-session interaction in both nucleus accumbens (F(1,40) = 4.4, p = 0.041) and caudate (F(1,40) = 4.8, p = 0.034). No interaction was detected in putamen, narrowing subsequent analyses to caudate and accumbens.1
The direction matters. In patients, both regions showed declining kicer from psychosis to remission. Pairwise post hoc tests:
- Caudate longitudinal decrease in patients: significant (t = 1.8, p = 0.043, one-sided test based on a priori hypothesis).
- Accumbens longitudinal decrease in patients: trend-level (t = 1.5, p = 0.065).
- Controls showed no longitudinal change in either region (full results in supplementary).
Cross-sectionally, patients during psychosis didn’t differ from controls at session 1. The differences emerged at session 2, during remission:1
- Accumbens during remission: patients lower than controls (t = 2.4, p = 0.01).
- Caudate during remission: patients lower than controls (t = 2.1, p = 0.02).
This is the clinically interesting finding. Standard dopamine-hypothesis framing predicts elevated striatal synthesis during psychosis and normalization during remission. What Schulz observed is partial undershoot — remitted patients had lower dopamine synthesis than healthy controls, not just normalized values. Acute-psychosis values weren’t elevated relative to controls in this cohort either, complicating the cross-sectional template that earlier meta-analyses built on.
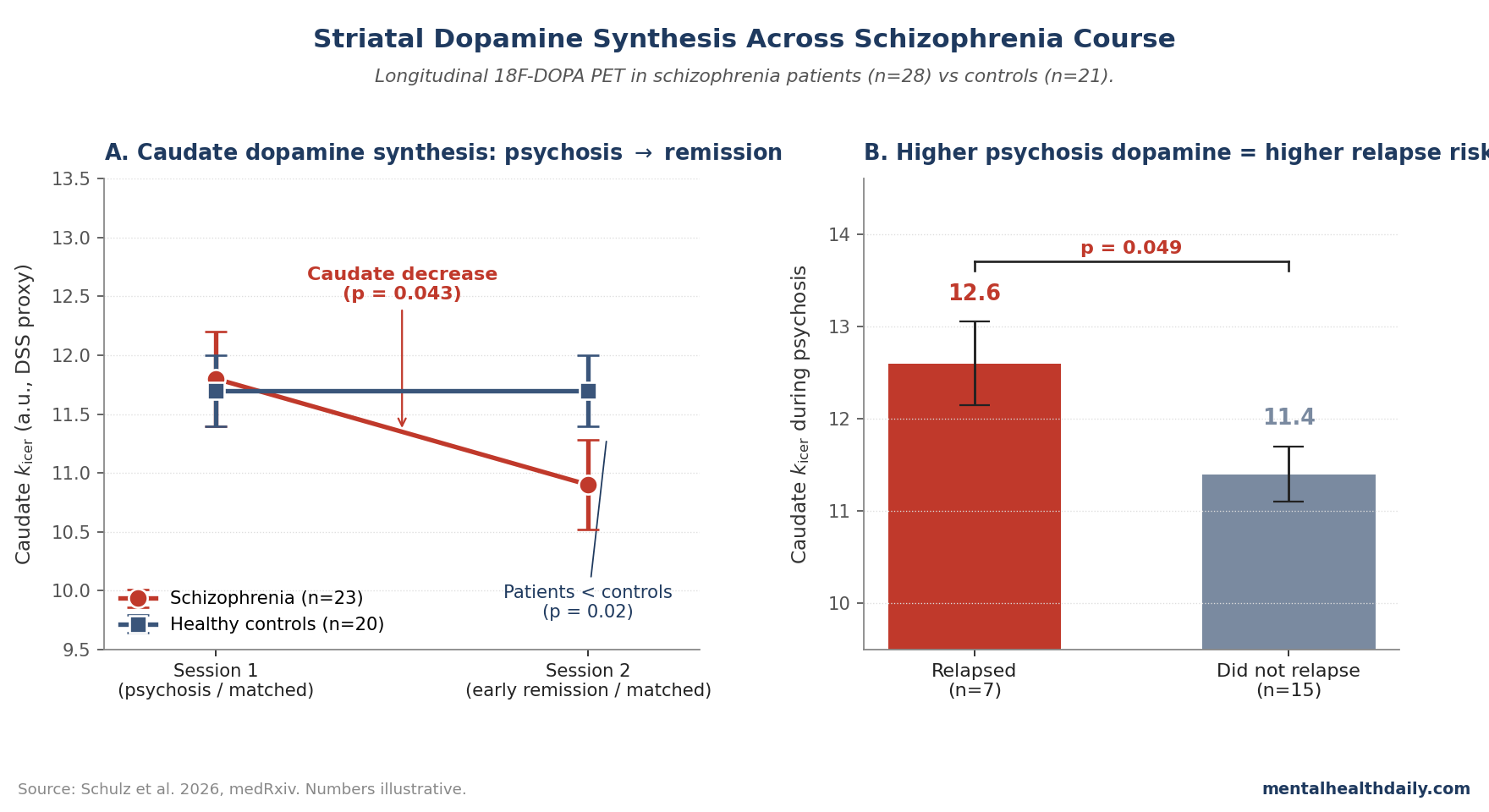
Caudate Dopamine During Psychosis Predicts Relapse
The exploratory follow-up at 12 months after session 2 found that 7 of 22 patients (32%) had experienced a psychotic relapse, consistent with the well-replicated 25–30% one-year relapse rate even on continued antipsychotic treatment.4
ANCOVAs comparing relapsing vs. non-relapsing patients (controlling for between-scan interval) found:1
- Caudate kicer during psychosis was higher in patients who later relapsed (F(1,19) = 4.4, p = 0.049).
- No difference in caudate during remission between relapsing and non-relapsing patients (p = 0.45).
- No differences in accumbens at either timepoint.
The pattern argues that the magnitude of presynaptic dopamine elevation during the acute episode — not the absolute value at remission — carries prognostic information about who will relapse. Patients with higher caudate synthesis during psychosis appeared to also have a slower initial recovery (between-scan intervals were longer in relapsers), and they were more likely to experience another episode within a year.
This is consistent with broader work on dopamine-hypothesis predictors of treatment response. Demjaha 2014 and 2017 found that treatment-resistant schizophrenia patients had lower baseline striatal dopamine synthesis than treatment-responsive patients,5 arguing that high-dopamine subtypes are the ones that respond well to D2-blocking antipsychotics. The Schulz finding flips a related question: among patients who do remit, those with the highest within-episode dopamine elevation may be most prone to recurrence.
What This Reframes About the Dopamine Hypothesis
Three implications follow.
- Dopamine synthesis is state-dependent within individuals. Most prior 18F-DOPA evidence has been cross-sectional, comparing different patients to different controls at single timepoints. The Schulz design is one of the first to track the same individuals across psychotic and remitted states, and it shows that kicer is dynamic over weeks-to-months — a timescale faster than aging-related dopamine declines but slower than the seconds-to-minutes phasic dopamine signals tied to reward learning. The mechanism for this medium-timescale modulation isn’t well understood. Candidates include D2 autoreceptor regulation, density of dopaminergic neuronal fibers, and functional connectivity within cortico-striatal-pallido-thalamic circuits.6
- The “more dopamine = more psychosis” framing is incomplete. Acute psychosis in this cohort wasn’t accompanied by elevation relative to healthy controls; remission was accompanied by undershoot. McCutcheon and colleagues’ 2018 network meta-analysis of 18F-DOPA studies found pooled effect sizes around d = 0.79 for striatal DSS in psychosis,2 but the heterogeneity is high (I² values often above 70%) and not all individual studies reproduce the elevation. Schulz adds another data point to the heterogeneity literature: in some patient samples, the cross-sectional contrast at any single timepoint may underrepresent the within-person dynamic.
- The remission undershoot has clinical implications. If patients in early remission have lower-than-control dopamine synthesis, sustained D2 blockade with antipsychotics might be operating against an already reduced presynaptic pool. This could partly explain the negative-symptom and cognitive complications that often persist or emerge during maintenance treatment, and it complicates the standard “minimum effective antipsychotic dose” question. The maintenance literature has shown that some patients can be successfully tapered7; the Schulz data raise the possibility that the patients with the lowest remission dopamine synthesis are different in kind from those whose synthesis normalizes.
How This Fits With Older Cross-Sectional Evidence
The dopamine hypothesis evolved in stages. The 1960s and 1970s framed schizophrenia as a fundamental excess of dopamine, motivated by the antipsychotic effects of D2-blocking drugs and the psychotomimetic effects of dopamine releasers like amphetamine. The 1990s introduced Davis and colleagues’ subdivided framework: hyperdopaminergia in mesolimbic projections (driving positive symptoms), hypodopaminergia in mesocortical projections (driving negative and cognitive symptoms).8
The Howes-Kapur 2009 reformulation refined the localization: 18F-DOPA studies showed elevated synthesis specifically in dorsal (associative) striatum — not the limbic ventral striatum that earlier reward-circuit models had emphasized.3 Subsequent imaging of the prodromal period (people at clinical high risk for psychosis) found that elevated striatal dopamine synthesis was already present before first-episode psychosis,9 consistent with dopamine elevation as a precursor rather than a consequence of the illness.
The most influential meta-analysis is McCutcheon 2018, pooling 18F-DOPA studies across schizophrenia, prodromal cohorts, and other psychiatric conditions.2 The aggregated finding: striatal DSS is elevated in schizophrenia (large pooled effect), with the strongest effects in dorsal striatum during active illness. Bipolar disorder, major depression, and OCD didn’t show the same striatal pattern, supporting the specificity of the dopamine signal to schizophrenia spectrum disorders.
Where Schulz adds value: the cross-sectional template doesn’t fully predict the longitudinal within-person dynamic. Patients can move from elevated (relative to their own remission baseline) during psychosis to lower-than-control during remission, and the magnitude of the within-person fluctuation may be more clinically informative than the absolute value at any one timepoint.
Limitations of the Schulz Synthesis
Small sample. 28 patients is well-powered for the within-person longitudinal interaction (the headline analysis) but is at the smaller end for cross-sectional group comparisons. The remission-undershoot finding (patients lower than controls during remission) carries a cross-sectional comparison and would benefit from larger replication.
Antipsychotic exposure is an inherent confound. All patients were medicated. The control analyses tested whether CPZ equivalents and antipsychotic plasma levels accounted for the longitudinal effect (they didn’t), and whether specific antipsychotics shifted the result (they didn’t). This isn’t a definitive ruling-out: chronic D2 blockade is known to affect presynaptic dopamine via long-loop autoregulation, and the longitudinal design partially captures this if dose-related medication effects shift with the patient’s clinical state.
Early remission specifically. The second scan was 6 weeks to 12 months after the first. This window was chosen specifically because most prior 18F-DOPA studies of remitted patients used much longer remission durations (often years), in which chronic dopaminergic deficits could obscure the dynamic shift Schulz wanted to capture. The trade-off is that “early remission” is a specific clinical state, and the findings may not generalize to long-term sustained remission.
Relapse follow-up was exploratory. The 12-month relapse analysis was not pre-specified as a primary endpoint, and only 22 of 28 patients were followed. The 32% relapse rate is consistent with literature, but the relapse-prediction finding (caudate kicer during psychosis predicting relapse) needs prospective confirmation in a pre-specified design.
The cross-sectional psychosis-vs-control comparison was non-significant. The standard hypothesis (elevated striatal dopamine synthesis during active psychosis) didn’t reach significance in this cohort. Schulz attributes this partly to sample size and partly to clinical heterogeneity; it’s also a reminder that not every individual patient sample reproduces the pooled meta-analytic effect.
Practical Implications for Schizophrenia Treatment
The Schulz findings are mechanistic rather than directly clinical, but two implications follow.
- Maintenance antipsychotic treatment operates against a moving target. If presynaptic dopamine synthesis fluctuates with psychotic-symptom state, then steady-dose D2 blockade is over-treating during remission and may be appropriately calibrated only during active episodes. This argues for the broader research literature on dose-reduction trials in stable patients,7 while flagging that abrupt discontinuation produces relapse and is well-documented to do so.10
- Relapse prediction may be possible from biomarkers measured during the acute episode rather than during remission. If higher caudate dopamine synthesis during psychosis is genuinely associated with downstream relapse, then 18F-DOPA imaging during the index admission could in principle be used to stratify patients for more intensive monitoring or longer-duration prophylactic treatment. This is far from clinical-translation-ready — PET imaging is expensive, the Schulz finding is from 22 patients, and effect sizes are modest — but it points at a research direction that could eventually yield individualized treatment-duration recommendations.
For most schizophrenia patients, the standard guidance still holds: continued antipsychotic treatment for at least 12 months after first remission, longer with multiple prior episodes, with structured planning for any dose-reduction trials. The Schulz data don’t change that practical calculus — they refine the underlying mechanistic story.
Common Questions About Dopamine and Schizophrenia
Is the dopamine hypothesis of schizophrenia still considered correct?
In its modern form, yes — though the framing has narrowed substantially since the 1970s. Current evidence supports elevated presynaptic dopamine synthesis in dorsal striatum during active psychosis (Howes-Kapur 2009 reformulation),3 with no convincing evidence for postsynaptic D2 receptor density elevation. Other neurotransmitter systems — particularly glutamate (NMDA receptor hypofunction) and GABA — are also implicated, and current models view dopamine as the final common pathway through which upstream disturbances produce psychotic symptoms.11
Why doesn’t every schizophrenia patient have elevated striatal dopamine?
Treatment-resistant schizophrenia (the ~30% of patients who don’t respond to D2-blocking antipsychotics) appears to have a different dopamine profile. Demjaha and colleagues’ 18F-DOPA work showed that treatment-resistant patients had similar or lower striatal dopamine synthesis than healthy controls, while treatment-responsive patients had clear elevation.5 This is consistent with treatment resistance involving non-dopaminergic mechanisms (likely glutamatergic) and is one reason clozapine — with its broader receptor profile — outperforms standard antipsychotics in this subgroup.
Does taking antipsychotics permanently change dopamine?
Long-term D2 blockade produces measurable adaptations in dopamine synthesis, receptor density, and downstream signaling. The clinical relevance of dopamine supersensitivity psychosis — the hypothesis that long-term antipsychotic exposure increases relapse risk on discontinuation — has been actively debated.12 The Schulz design partially controls for this by measuring patients on stable medication regimens at both timepoints, but it doesn’t address what happens after long-term exposure.
Can dopamine PET predict which patients will relapse?
Tentatively, in research settings. The Schulz exploratory analysis found that higher caudate dopamine synthesis during the acute psychotic episode was associated with relapse within 12 months (F = 4.4, p = 0.049, n = 22).1 This needs prospective confirmation in a pre-specified design before any clinical application. Clinical-grade biomarkers for relapse prediction don’t currently exist; symptomatic monitoring and medication adherence remain the practical levers.
What about the glutamate hypothesis?
The NMDA-receptor-hypofunction hypothesis (Olney-Farber, then Coyle) is the leading non-dopaminergic framework. NMDA antagonists like ketamine and PCP produce schizophrenia-like positive, negative, and cognitive symptoms in healthy volunteers, and 1H-MRS studies have shown altered glutamate levels in anterior cingulate and other regions in unmedicated patients.11 Current models don’t oppose glutamate to dopamine but instead position glutamatergic dysfunction upstream of the dopamine signal, with cortical-striatal disinhibition driving the presynaptic dopamine elevation in psychosis.
How should I think about the relapse risk in someone who’s recovered from a psychotic episode?
The 12-month relapse rate after a first psychotic episode is roughly 25–30% on continued antipsychotic treatment, and 70–80% off treatment.10 Even within the medicated group, relapse is the modal expected outcome over a 5-year horizon for most patients. The practical implication is that “well now” doesn’t mean “low ongoing risk” — structured monitoring, medication adherence support, and early-warning-sign planning matter even in stable remission.
References
- Striatal dopamine synthesis in schizophrenia decreases from psychosis to psychotic remission. Schulz J et al. medRxiv. 2026 (preprint). doi:10.64898/2026.04.20.26351256
- Schizophrenia, dopamine and the striatum: from biology to symptoms. McCutcheon RA et al. Trends in Neurosciences. 2019;42(3):205-220. doi:10.1016/j.tins.2018.12.004
- The dopamine hypothesis of schizophrenia: version III – the final common pathway. Howes OD, Kapur S. Schizophrenia Bulletin. 2009;35(3):549-562. doi:10.1093/schbul/sbp006
- Maintenance treatment with antipsychotic drugs for schizophrenia. Leucht S et al. Cochrane Database of Systematic Reviews. 2012;5:CD008016. doi:10.1002/14651858.CD008016.pub2
- Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. Demjaha A et al. American Journal of Psychiatry. 2012;169(11):1203-1210. doi:10.1176/appi.ajp.2012.12010144
- Dopaminergic regulation of striatal microcircuits. Cox J, Witten IB. Nature Reviews Neuroscience. 2019;20(8):482-494. doi:10.1038/s41583-019-0189-2
- Dose reduction or discontinuation of antipsychotics in patients with schizophrenia and a history of psychosis: a systematic review. Bogers JPAM et al. Schizophrenia Bulletin. 2020;46(6):1399-1410. doi:10.1093/schbul/sbaa097
- Dopamine in schizophrenia: a review and reconceptualization. Davis KL et al. American Journal of Psychiatry. 1991;148(11):1474-1486. doi:10.1176/ajp.148.11.1474
- Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Howes OD et al. Archives of General Psychiatry. 2009;66(1):13-20. doi:10.1001/archgenpsychiatry.2008.514
- Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Robinson D et al. Archives of General Psychiatry. 1999;56(3):241-247. doi:10.1001/archpsyc.56.3.241
- The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Hu W et al. Annals of the New York Academy of Sciences. 2015;1338:38-57. doi:10.1111/nyas.12547
- Antipsychotic-induced dopamine supersensitivity psychosis: pharmacology, criteria, and therapy. Chouinard G et al. Psychotherapy and Psychosomatics. 2017;86(4):189-219. doi:10.1159/000477313