
A 2026 Nature Neuroscience study found that lecanemab cleared amyloid in a humanized-microglia Alzheimer’s disease model only when microglia and the antibody’s intact Fc fragment were both present: X-34 plaque area differed across IgG1, lecanemab, and Fc-silenced LALA-PG groups (P = 0.0003), 82E1 plaque area differed across groups (P < 0.0001), and microglia-deficient mice showed no plaque-clearance effect.1
Research Highlights
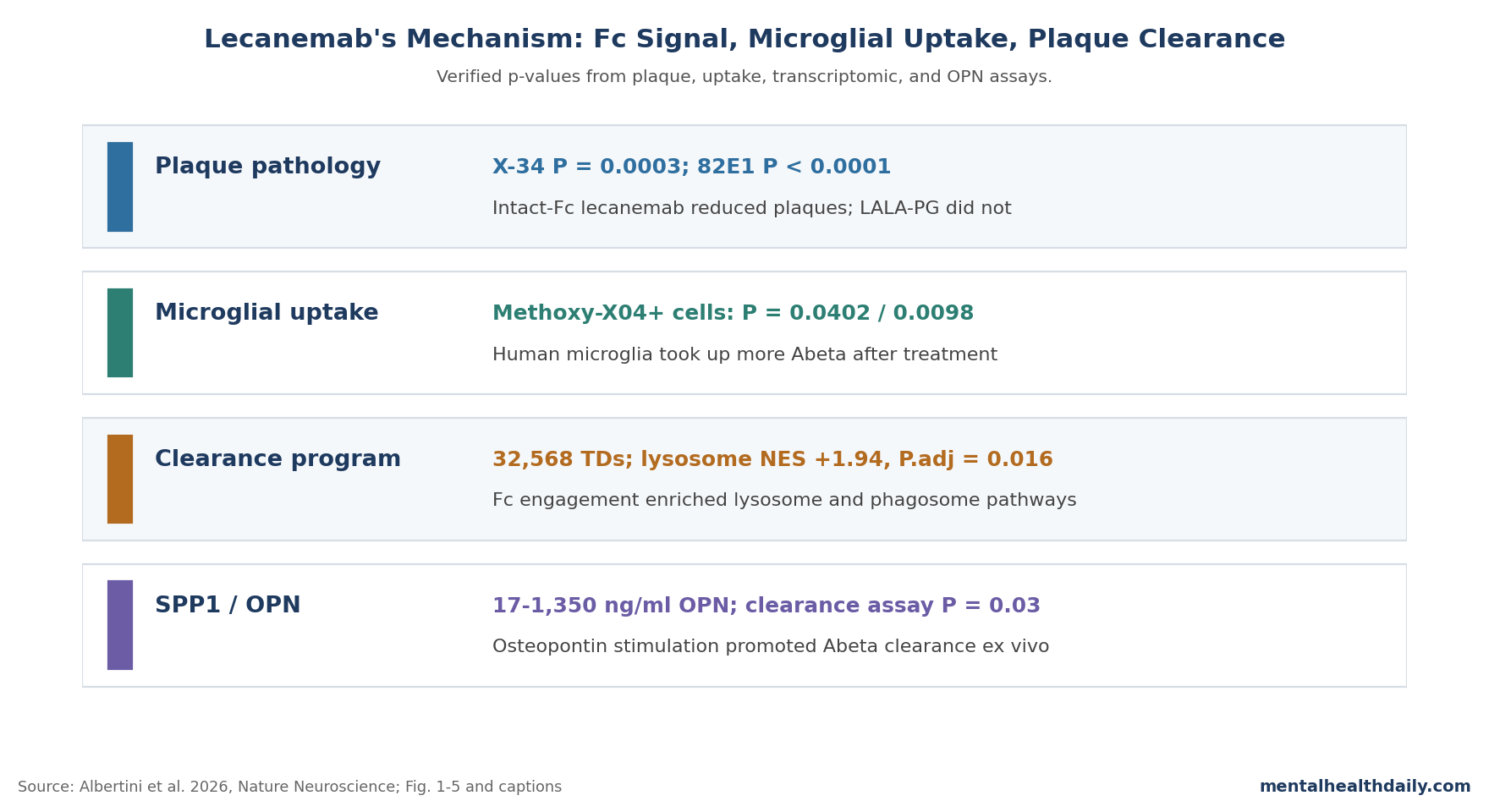
- Fc engagement was required: standard lecanemab reduced X-34 and 82E1 plaque measures after 8 weeks, while Fc-silenced LALA-PG did not despite intact plaque binding (X-34 P = 0.0003; 82E1 P < 0.0001).1
- Microglia were not optional: lecanemab failed to clear plaques in mice genetically lacking microglia, and human microglia showed higher Methoxy-X04 amyloid uptake after treatment (P = 0.0402; CD68-high subset P = 0.0098).1
- The transcriptomic signal was targeted: spatial transcriptomics analyzed 32,568 cortical tissue domains; lysosome genes were enriched near plaques in lecanemab-treated tissue domains (NES +1.94; adjusted P = 0.016).1
- SPP1/osteopontin became a functional clue: OPN rose around plaques (P = 0.0008 and P = 0.0007 across X-34 and 82E1 regions), and 1,350 ng/ml OPN reduced 82E1 plaque coverage in an ex vivo clearance assay (P = 0.03).1
- The clinical claim stays narrower: CLARITY-AD reported about 27% slowing of decline over 18 months, but Albertini et al. tested plaque-clearance mechanism, not patient cognition or ARIA risk prediction.2
Lecanemab is an anti-amyloid monoclonal antibody for early symptomatic Alzheimer’s disease. It binds amyloid-beta aggregates and, in clinical trials, lowers amyloid plaque burden while producing a modest average slowing of cognitive and functional decline.2
The disputed part has been the mechanism. One model treats lecanemab as a plaque-binding antibody that passively disrupts amyloid deposits.
Another model says the antibody’s Fc region — the tail of the antibody that immune cells can grab — recruits microglia to engulf and degrade antibody-coated amyloid. Albertini et al. tested that second model directly by separating plaque binding from Fc signaling.
Fc-Silenced Lecanemab Bound Plaques But Failed to Clear Them
The core experiment used AppNL-G-F Csf1rΔFIRE/ΔFIRE mice, an amyloid-pathology model engineered to lack endogenous mouse microglia, then xenotransplanted with human induced-pluripotent-stem-cell-derived microglia. The setup let researchers test a human antibody against human microglia inside an amyloid-loaded mouse brain.
The treatment comparison was unusually clean. Standard lecanemab kept its Fc effector function.
Lecanemab LALA-PG kept the same plaque-binding region but carried LALA-PG mutations that silence Fc-mediated immune engagement. Both antibodies could bind amyloid plaques, but only intact-Fc lecanemab reduced plaque pathology.
After 8 weeks of 10 mg kg−1 weekly treatment, X-34 plaque area differed significantly across IgG1, lecanemab, and LALA-PG groups (P = 0.0003; IgG1 n = 10, lecanemab n = 12, LALA-PG n = 9). 82E1 plaque area, which marks amyloid-beta peptides more broadly, also differed across groups (P < 0.0001).
Pairwise p-values in the figure placed lecanemab below both IgG1 and LALA-PG for plaque burden, while IgG1 and LALA-PG were not the active-clearance pair.1
That result matters because the LALA-PG antibody was not absent from plaques. The paper reports strong plaque accumulation of LALA-PG, including after 2 weeks.
Plaque binding alone therefore was not enough. The Fc fragment had to communicate with microglia.
Microglia Were Required for the Amyloid-Clearing Effect
Albertini et al. also tested the reverse side of the mechanism. If intact-Fc lecanemab clears plaques by recruiting microglia, then removing microglia should blunt the effect even when the antibody itself is unchanged.
That is what happened. AppNL-G-F Csf1rΔFIRE/ΔFIRE mice without human microglia xenotransplantation did not show plaque-load improvement with standard lecanemab.
The antibody could exist in the system, but the immune effector cell needed to process antibody-coated plaque was missing.
The same mechanism appeared in functional uptake assays. In mice with human microglia, lecanemab increased the percentage of Methoxy-X04-positive hCD45-positive microglia (P = 0.0402; IgG1 n = 8, lecanemab n = 7).
The CD68-high microglial subset — cells with a more phagolysosomal, debris-processing phenotype — showed a stronger uptake signal (P = 0.0098).1
Those uptake data keep the interpretation grounded. The study does more than show that plaques were lower after treatment.
It shows a plausible intermediate step: microglia physically contained more amyloid after the antibody exposed plaques to Fc-mediated clearance.
Lecanemab Activated Lysosome, Phagosome, and Antigen-Presentation Genes
Microglial activation can mean many things, including helpful clearance, inflammatory injury, or broad immune noise. Albertini et al. used spatial transcriptomics and single-cell RNA sequencing to ask what kind of activation lecanemab produced near amyloid plaques.
The spatial dataset included 32,568 cortical tissue domains from 4 samples: 17,186 domains from lecanemab-treated samples and 15,382 from LALA-PG-treated samples. Each domain was a 40-μm hexbin pseudospot, with an average of 61.3 human genes and 74.4 unique molecular identifiers.
Human-derived reads could be assigned to xenografted microglia because the host tissue was mouse-derived.1
Near plaques, lecanemab-treated tissue domains showed upregulation of genes linked to lysosomal function, phagocytosis, antigen presentation, metabolism, and interferon-responsive programs. The source numbers are sharper than a generic “immune activation” summary:
- Lysosome pathway: NES +1.94, adjusted P = 0.016 in lecanemab tissue domains near pathology.
- Phagosome pathway: NES +1.57, adjusted P = 0.027 in lecanemab tissue domains near pathology.
- Single-cell RNA sequencing: 22,420 human microglial cells after macrophage removal, including 10,850 IgG1 cells and 11,570 lecanemab cells.
- Differential expression: 126 genes were significantly upregulated and 166 were significantly downregulated after lecanemab treatment at adjusted P < 0.05.
The LALA-PG arm is the calibration point. Fc-silenced antibody accumulated on plaques but did not induce the same clearance-associated program.
That makes Fc engagement the likely driver of the transcriptional pattern, because antibody binding to amyloid without Fc signaling did not produce the same response.
SPP1/Osteopontin Linked Microglial State to Plaque Removal
SPP1 is the gene encoding osteopontin, a secreted protein involved in immune-cell activation, migration, and tissue remodeling. In this study, SPP1 was more than a label on an activated microglial state.
It became a plausible effector in the plaque-clearance chain.
Albertini et al. report that SPP1 was the most strongly upregulated gene in both single-cell RNA sequencing and spatial differential-expression analyses. Weighted gene coexpression network analysis placed SPP1 in a pink module enriched around amyloid plaques in lecanemab-treated tissue domains.
Confocal imaging then showed higher OPN-positive area within IBA1-positive microglia around X-34-positive and 82E1-positive plaques (P = 0.0008 and P = 0.0007).1
The functional assay is the important step. Human-derived microglia were plated onto 6-month-old AppNL-G-F brain cryosections and stimulated with human OPN concentrations from 17 to 1,350 ng ml−1.
At the highest concentration tested, OPN reduced 82E1 plaque coverage relative to no OPN (P = 0.03; n = 3 independent experiments).1
That does not prove that blood or cerebrospinal-fluid SPP1 can already guide lecanemab decisions in patients. It does make SPP1/osteopontin a mechanistically credible response marker to measure in trials, especially if future work can show that its induction tracks amyloid PET change, ARIA risk, or clinical trajectory.
Fc Biology Also Explains Why Anti-Amyloid Risk Is Hard to Separate From Benefit
Fc engagement is not automatically good. Sun et al. reported that Fc effector activity of anti-amyloid antibody could drive synapse loss and cognitive deficits in an Alzheimer’s-like mouse model.5
Jung et al. took a different route by testing a Gas6 fusion strategy meant to promote amyloid-beta clearance through a less inflammatory phagocytic pathway.6
Albertini et al. did not see the same broad pro-inflammatory pattern emphasized in some other antibody work. Lecanemab-treated human microglia upregulated interferon-responsive genes but did not broadly induce classic inflammatory cytokines such as TNF and IL1B.
Synaptophysin and synaptic puncta around plaques were unchanged, and Homer1 showed a small increase rather than a loss signal.1
The safety limit is still substantial. The model has reduced ability to capture vascular pathology or blood-brain barrier dysfunction, and amyloid-related imaging abnormalities in patients may involve vessel-wall amyloid, border-associated macrophages, complement, APOE genotype, and vascular fragility.
CLARITY-AD remains the patient-level anchor: lecanemab slowed decline by about 27% over 18 months, but ARIA occurred more often in the active-treatment group than in the placebo group.2
Mechanism Is Stronger Than the Clinical Inference
The Albertini paper is strong mechanistic evidence for lecanemab’s plaque-clearance pathway. It is not a new clinical efficacy trial, and it should not be read as proof that increasing microglial activation will improve memory in patients.
What the model can support: in the tested amyloid mouse systems, lecanemab requires Fc-mediated microglial effector function for robust plaque removal. The convergence of Fc-silenced antibody failure, microglia-deficient failure, microglial uptake, transcriptomic activation, and OPN-linked clearance makes passive plaque dissolving a weak explanation.
What the model cannot support: direct claims about which patients will benefit, whether SPP1 should be used clinically, or how to engineer the best efficacy-to-ARIA ratio. The xenograft model is valuable because it uses human microglia and a human antibody, but it does not reproduce the full immune, vascular, and clinical environment of a person with Alzheimer’s disease.
The practical implication is a design constraint for next-generation antibodies. If Fc signaling is necessary for plaque clearance, simply removing Fc effector function may protect against some immune risk while also weakening the clearance mechanism.
Better engineering would need to tune microglial phagocytosis, complement activation, and vascular safety together, not pretend one knob controls the whole system.
Questions About Lecanemab, Microglia, and Fc Receptors
Why did Fc-silenced lecanemab fail if it still bound plaques?
Plaque binding placed the antibody on amyloid, but LALA-PG mutations silenced the Fc signal that recruits immune effector functions. In the Albertini study, that left antibody stuck on plaques without producing the microglial uptake and lysosomal clearance program seen with intact-Fc lecanemab.1
Does this prove lecanemab helps cognition by activating microglia?
No. It supports a mechanism for plaque removal in model systems.
Cognitive slowing in patients still comes from clinical trials such as CLARITY-AD, where the average benefit was modest even though amyloid lowering was substantial.2
Could stronger Fc activation make lecanemab work better?
Possibly, but the safety side of the same pathway is the problem. Fc engagement can promote phagocytosis, yet related immune mechanisms may also contribute to synaptic injury or vascular complications in some contexts.
Future antibodies need better-tuned effector function, not maximal immune activation by default.5
Should SPP1 or osteopontin be treated as a patient biomarker now?
Not yet. The 2026 study showed SPP1 induction near plaques and OPN-driven amyloid clearance in an ex vivo assay.
A clinical biomarker would need human longitudinal evidence linking SPP1 or OPN changes to amyloid PET, ARIA, cognition, or treatment durability.
Does this mechanism apply to donanemab or aducanumab too?
The broad principle probably applies to other plaque-binding anti-amyloid antibodies, but the exact immune program may differ by antibody target, Fc structure, dose, and plaque species. Aducanumab and donanemab both lower amyloid plaques clinically, but Albertini et al. specifically tested lecanemab and mouse analog mAb158.3,4
References
- The Alzheimer’s therapeutic Lecanemab attenuates Aβ pathology by inducing an amyloid-clearing program in microglia. Albertini G et al. Nature Neuroscience. 2026;29:100–110. doi:10.1038/s41593-025-02125-8
- Lecanemab in early Alzheimer’s disease. Van Dyck CH et al. New England Journal of Medicine. 2023;388(1):9–21. doi:10.1056/NEJMoa2212948
- The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Sevigny J et al. Nature. 2016;537:50–56. doi:10.1038/nature19323
- Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. Sims JR et al. JAMA. 2023;330(6):512–527. doi:10.1001/jama.2023.21109
- Fc effector of anti-Aβ antibody induces synapse loss and cognitive deficits in Alzheimer’s disease-like mouse model. Sun X-Y et al. Signal Transduction and Targeted Therapy. 2023;8:30. doi:10.1038/s41392-023-01350-6
- Anti-inflammatory clearance of amyloid-β by a chimeric Gas6 fusion protein. Jung H et al. Nature Medicine. 2022;28:1802–1812. doi:10.1038/s41591-022-01926-9
- Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related amyloid-β pathology. Mancuso R et al. Nature Neuroscience. 2024;27:886–900. doi:10.1038/s41593-024-01600-y