
GLP-1 drugs are usually described as appetite drugs, but a 2026 Nature mouse study mapped a more specific reward circuit: small-molecule GLP-1 receptor agonists suppressed palatable-food intake through Glp1r-expressing central-amygdala neurons that reduced dopamine release in the nucleus accumbens during high-fat-food retrieval.1 That mechanism makes binge-eating and substance-use hypotheses more plausible without turning an animal circuit study into a human treatment trial.
Research Highlights
- Reward feeding was separable: A single S33W receptor substitution let mice respond to human-selective small-molecule GLP-1 drugs, and the drugs reduced both standard-diet and high-fat-diet intake across multiple cohorts.1
- Behavior separated from nausea: Machine-learning home-cage profiling found 28 LiCl-sensitive, 25 liraglutide-sensitive, 22 danuglipron-sensitive, and 13 orforglipron-sensitive behavioral syllables, with orforglipron clustering away from the LiCl nausea reference.1
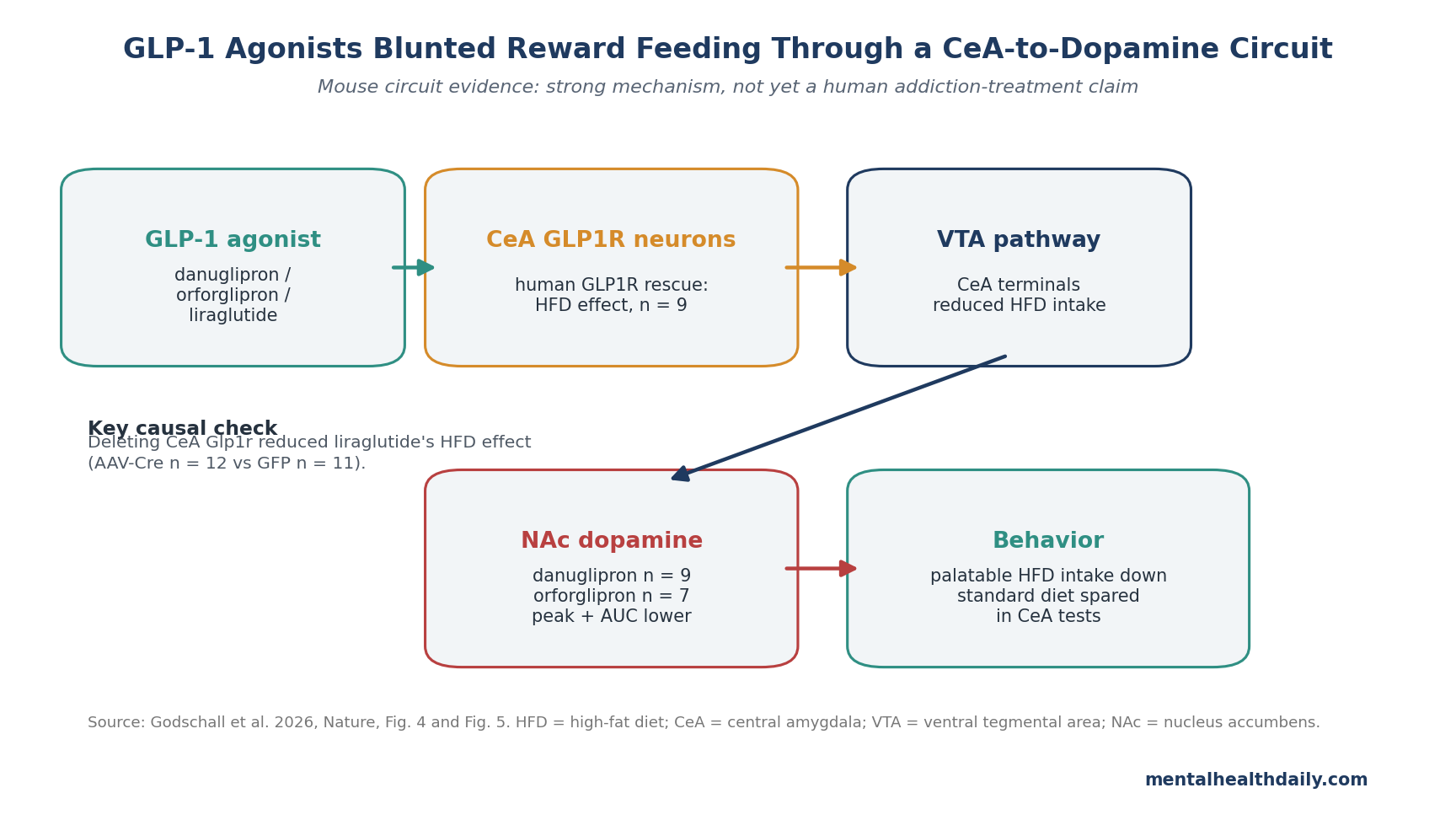
- Central amygdala was causal: Expressing human GLP1R in central-amygdala Glp1r neurons (n = 9) let danuglipron suppress high-fat-diet intake, while deleting CeA Glp1r reduced liraglutide’s high-fat-diet effect in AAV-Cre mice (n = 12) vs. GFP controls (n = 11).1
- Dopamine dropped during reward retrieval: Nucleus-accumbens dopamine photometry showed that danuglipron (n = 9) and orforglipron (n = 7) reduced high-fat-food-evoked dopamine peak and area-under-the-curve signals.1
- Addiction relevance is still indirect: Real-world semaglutide studies reported lower alcohol-use-disorder and cannabis-use-disorder incidence in 2024 cohorts, but the mouse circuit does not prove that GLP-1 drugs treat addiction or binge eating in people.2,3
GLP-1 receptor agonists are drugs that activate the glucagon-like peptide-1 receptor, a hormone-sensitive receptor involved in insulin release, satiety, and brain control of feeding. Semaglutide and liraglutide are peptide drugs; newer agents such as danuglipron and orforglipron are smaller non-peptide molecules designed for oral use.
Godschall et al. were not testing weight loss in patients. They were asking how small-molecule GLP-1 agonists reach reward-driven feeding circuits when ordinary rodent receptors do not respond to many of the human-selective compounds.
Humanized GLP1R Mice Made Small-Molecule Testing Possible
Many non-peptide GLP-1 agonists bind human GLP1R differently than mouse GLP1R because of a single amino-acid difference at receptor position 33. The researchers used CRISPR-Cas9 genome editing to replace the mouse serine with the human tryptophan, creating Glp1rS33W mice that kept normal baseline metabolic function but gained sensitivity to danuglipron and orforglipron.1
Why this model matters: without the S33W substitution, danuglipron and orforglipron would look weak or inactive in ordinary mouse experiments, not because the reward circuit is irrelevant, but because the receptor pharmacology is mismatched. The model let the study test a drug class that is clinically aimed at humans while still using mouse circuit tools.
After validation, the drugs reduced active-phase standard-diet intake and rest-phase high-fat-diet intake. Standard-diet intake is closer to hunger-driven feeding. High-fat-diet intake during a normally low-feeding phase is a reward-feeding assay: the food is palatable enough that mice eat it when they otherwise would eat little.
Home-Cage Behavior Separated Satiety From Malaise
Nausea is a major clinical limitation of GLP-1 drugs, so any claim about reward control has to avoid a simple interpretation: mice ate less because they felt sick. The study used conditioned taste avoidance, open-field behavior, elevated-plus-maze behavior, and 2-hour home-cage video tracking to separate satiety-like patterns from malaise-like patterns.1
Behavioral profiling: pose-estimation and Keypoint-MoSeq analysis identified 91 behavioral syllables. LiCl, the nausea reference, significantly changed 28 syllables. Liraglutide changed 25, danuglipron changed 22, and orforglipron changed 13.
That count is not a clinical adverse-event scale, but it shows why the circuit work is more than a generic “the animals stopped eating” result. Orforglipron in particular maintained a more active, exploratory pattern and separated from the LiCl nausea reference in principal-component analysis. Danuglipron and liraglutide sat closer to satiety or malaise-like behavioral space.
Central-Amygdala GLP1R Neurons Suppressed High-Fat Food Intake
Central amygdala (CeA) refers to an amygdala subregion involved in threat, valence, feeding, and motivated behavior. The 2026 study found that danuglipron and orforglipron increased FOS activity, a marker of recent neuronal activation, in the nucleus tractus solitarius, area postrema, and CeA of humanized Glp1r mice.1
The causal tests were tighter than the activation map. When the researchers expressed human GLP1R only in Glp1r-expressing CeA neurons, danuglipron suppressed high-fat-diet intake without the same effect on standard-diet intake. In other words, activating the CeA node was enough to reproduce the reward-feeding component of the drug effect.
Deletion pushed the other direction. Removing Glp1r from CeA neurons reduced liraglutide’s ability to inhibit high-fat-diet intake while leaving standard-diet intake largely intact. That makes the CeA node a necessary contributor to the palatable-food effect, beyond a region that lights up after systemic drug exposure.
The Amygdala-to-Dopamine Link Explains the Addiction Hypothesis
Nucleus accumbens dopamine is a reward-signal system often recruited by palatable food, addictive drugs, and cue-driven wanting. Godschall et al. used dLight photometry, a fluorescent dopamine-sensor method, to measure dopamine signals in the nucleus accumbens during high-fat-food retrieval.1
Danuglipron and orforglipron reduced both peak and area-under-the-curve dopamine signals during high-fat-food retrieval in Glp1rS33W mice. The effect was absent in wild-type mice for danuglipron, which fits the receptor-specific design: without the humanized receptor, the small molecule could not produce the same dopamine effect.
Tracing and optogenetic tests connected the CeA to the ventral tegmental area (VTA), a midbrain dopamine hub that projects to the nucleus accumbens. Stimulating CeA Glp1r axon terminals in the VTA reduced high-fat-diet intake without reducing standard-diet intake. In the simplest circuit language, the proposed pathway is:
- NTS input: preproglucagon neurons in the nucleus tractus solitarius can send GLP-1-related input to the CeA.
- CeA gate: Glp1r-expressing CeA neurons respond to pharmacological and endogenous GLP-1 signaling.
- VTA output: CeA projections influence midbrain dopamine circuitry.
- NAc signal: nucleus-accumbens dopamine during palatable-food retrieval is blunted.
- Behavior: high-fat-food intake falls more selectively than standard-diet intake.
This is why the paper naturally points toward binge eating and substance-use disorders. It identifies a drug-sensitive reward pathway with gut-brain satiety and mesolimbic-motivation components.
Human Addiction Signals Are Encouraging but Observational
The addiction claim did not begin with this mouse paper. Two 2024 real-world cohort analyses by Wang et al. reported that semaglutide exposure was associated with lower incidence and recurrence of alcohol use disorder and cannabis use disorder.2,3 Those studies were clinically interesting because they involved human health-record data alongside animal feeding assays.
But observational semaglutide studies have a different weakness: prescribing is not random. People who receive semaglutide can differ from comparator patients in obesity severity, diabetes care, clinical contact, motivation, insurance access, and unmeasured health behavior. Statistical adjustment helps, but it cannot turn routine-care prescribing into a randomized addiction trial.
Adjacent circuit work makes the pattern more coherent. Merkel et al. reported that an endogenous GLP-1 circuit engaged VTA GABA neurons to regulate mesolimbic dopamine neurons and attenuate cocaine seeking.4 Fortin and Roitman earlier linked central GLP-1 receptor activation to cocaine-evoked phasic dopamine signaling in the nucleus accumbens core.5
Together, the evidence supports a cautious thesis: GLP-1 signaling can reach reward circuitry relevant to food and drugs, and semaglutide-class drugs deserve disorder-specific trials. It does not support casual claims that GLP-1 drugs are already proven anti-addiction medications.
Limits of the Mouse Reward-Circuit Evidence
Animal model boundary: high-fat-food intake in mice is useful for isolating reward-feeding biology, but it is not binge-eating disorder, alcohol use disorder, cannabis use disorder, or cocaine use disorder. Compulsive human behavior includes withdrawal, cue exposure, social context, psychiatric comorbidity, dose escalation, and relapse risk that a 1-hour or 4-hour feeding assay cannot model.
Small intracranial cohorts: many of the strongest causal experiments used viral targeting, fiber implants, optogenetics, or photometry in small mouse cohorts. That is normal for circuit neuroscience, but it limits precision around effect size and generalizability.
Drug differences matter: liraglutide, semaglutide, danuglipron, and orforglipron are not interchangeable. They differ in molecular size, route, half-life, receptor binding, brain penetration, and adverse-effect profile. A circuit activated by one GLP-1 agonist does not automatically predict the same psychiatric effect for every drug in the class.
Clinical translation: if GLP-1 drugs reduce addictive behavior, the most plausible target may be reward salience, craving, or cue-triggered consumption rather than all-purpose “willpower.” That makes trial design important: alcohol, cannabis, stimulant, opioid, and binge-eating outcomes should not be collapsed into one generic reward endpoint.
Questions About GLP-1 Drugs and Reward Circuits
Does this prove semaglutide treats addiction?
No. The 2026 study mapped a mouse circuit for palatable-food intake, and 2024 observational studies linked semaglutide to lower alcohol- and cannabis-use-disorder outcomes. A treatment claim needs randomized human trials with disorder-specific endpoints.
Is the central amygdala a hunger center?
Not in the simple sense. In this study, CeA Glp1r neurons were more tied to reward-driven high-fat-food intake than to standard-diet intake, which makes them look more like a valence and motivation gate than a generic calorie detector.
Why does dopamine matter here?
Dopamine release in the nucleus accumbens helps encode reward pursuit and cue-driven wanting. Lower dopamine during palatable-food retrieval gives a mechanistic explanation for why a GLP-1 agonist might make highly rewarding intake less compelling.
Which clinical trials would be most informative?
Randomized trials in binge-eating disorder and specific substance-use disorders would be most informative, with craving, cue reactivity, actual consumption, relapse, adverse effects, and discontinuation tracked separately. Trials also need to compare specific drugs rather than treating GLP-1 agonists as one uniform psychiatric intervention.
References
- Godschall EN, Gungul TB, Sajonia IR, et al. A brain reward circuit inhibited by next-generation weight-loss drugs in mice. Nature. 2026. doi:10.1038/s41586-026-10444-4
- Wang W, et al. Associations of semaglutide with incidence and recurrence of alcohol use disorder in real-world population. Nature Communications. 2024;15:4548. PubMed
- Wang W, et al. Association of semaglutide with reduced incidence and relapse of cannabis use disorder in real-world populations: a retrospective cohort study. Molecular Psychiatry. 2024;29:2587-2598. PubMed
- Merkel R, et al. An endogenous GLP-1 circuit engages VTA GABA neurons to regulate mesolimbic dopamine neurons and attenuate cocaine seeking. Science Advances. 2025;11:eadr5051. PubMed
- Fortin SM, Roitman MF. Central GLP-1 receptor activation modulates cocaine-evoked phasic dopamine signaling in the nucleus accumbens core. Physiology & Behavior. 2017;176:17-25. PubMed