
A Cell Reports study tied C9orf72-related ALS/FTD inflammation to a gut-bacterial product: inflammatory microbial glycogen appeared in 15 of 22 ALS fecal samples, 1 of 1 C9orf72-frontotemporal dementia sample, and 4 of 12 healthy controls.1 The result supports a narrower gene-by-microbial-product mechanism that makes C9orf72 loss of function more inflammatory.
Research Highlights
- Human samples carried the signal: inflammatory glycogen was detected in 15 of 22 ALS samples and 4 of 12 healthy-control samples.1
- C9orf72 changed the immune response: C9orf72-deficient macrophages released more tumor necrosis factor after exposure to several gut bacteria, including Parabacteroides merdae.1
- Glycogen was modifiable: alpha-amylase digestion reduced inflammatory activity, and gut-targeted enzymatic treatment promoted survival in C9orf72-deficient mice.1
- The mechanism was more specific than dysbiosis: metatranscriptomics separated pro-inflammatory and pro-survival environments with an AUC of 0.98.1
- Translation stays cautious: the human fecal panel contained only 35 people, so the finding needs multi-site longitudinal ALS/FTD cohorts before it becomes a biomarker or intervention target.1
C9orf72 is a gene strongly linked to amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). ALS primarily damages motor neurons, while FTD affects behavior, language, and executive function; the 2 disorders overlap biologically, and C9orf72 repeat expansions are one of the best-established shared genetic causes.2
Microbial glycogen means branched glucose storage molecules produced by bacteria. The McCourt et al. study found that some gut-bacterial glycogen forms behaved like inflammatory ligands, especially when myeloid cells lacked normal C9orf72 function. Myeloid cells include macrophages and related innate immune cells that detect microbes and release cytokines.
15 of 22 ALS Samples Contained Inflammatory Microbial Glycogen
Researchers tested washed, heat-killed fecal bacteria from 35 people: 12 healthy controls, 22 patients with ALS, and 1 patient with C9orf72-FTD. They exposed macrophages to those preparations with or without alpha-amylase, an enzyme that digests glycogen, then measured tumor necrosis factor release.1
Human signal: alpha-amylase-sensitive inflammatory glycogen appeared in 20 of 35 fecal samples overall. Disease-stratified results were sharper: 15 of 22 ALS samples, 1 of 1 C9orf72-FTD sample, and 4 of 12 healthy-control samples showed this activity. Aggregate alpha-amylase-sensitive activity was enriched in ALS/FTD feces (p = 0.01) but not in healthy-control feces (p = 0.82).1
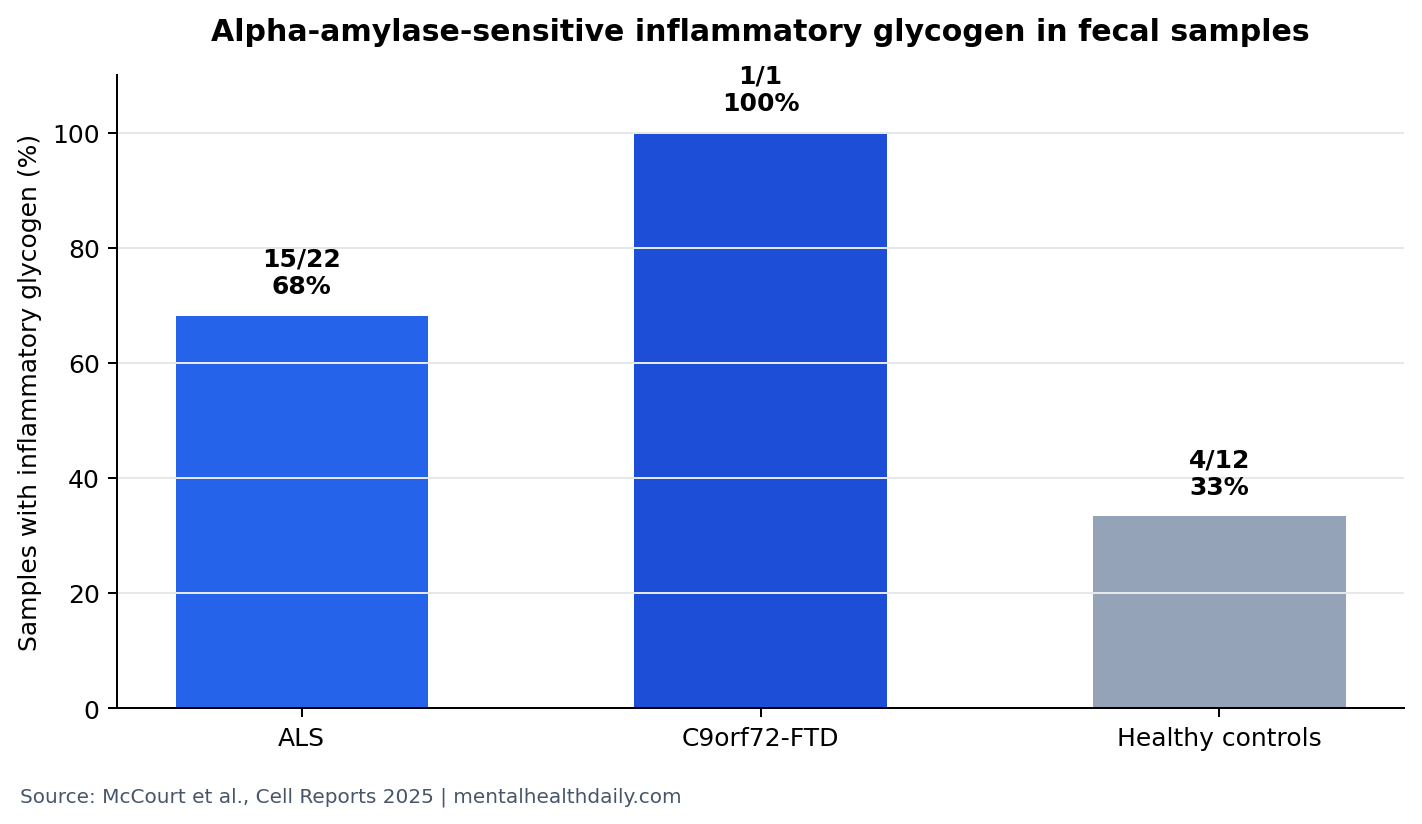
Plain-English interpretation: the human data suggest that a microbial product capable of provoking macrophage cytokine release is more common in ALS/FTD fecal samples than in controls. The sample is too small for diagnosis, but the direction fits the mouse and macrophage mechanism.
Parabacteroides merdae Turned a Genetic Risk State Into Inflammation
The study did not stop at sequencing lists of gut bacteria. Researchers exposed C9orf72-normal and C9orf72-deficient macrophages to fecal preparations and individual bacterial strains, then asked which exposures triggered cytokine release. Parabacteroides merdae was one of several strains that produced stronger tumor necrosis factor release from C9orf72-deficient macrophages than from C9orf72-normal cells.1
Tumor necrosis factor is an inflammatory cytokine, meaning a signaling protein that immune cells use to coordinate inflammation. In this paper, tumor necrosis factor served as the assay readout that let researchers sort microbial preparations into more inflammatory and less inflammatory groups.
Metatranscriptomic analysis then pointed away from a simple “bad species” story. The glycogen biosynthesis pathway separated pro-inflammatory from pro-survival microbial environments, and a classifier built from microbial transcripts reached an AUC of 0.98. AUC means area under the receiver operating curve; 0.50 is chance discrimination and 1.00 is perfect separation.
Alpha-Amylase Made the Mechanism More Than a Correlation
Alpha-amylase breaks down glycogen and related glucose polymers. When researchers treated bacterial preparations with alpha-amylase, C9orf72-deficient macrophages released less tumor necrosis factor. That enzymatic step targeted the proposed molecular driver directly, beyond the bacteria carrying it.1
Mouse experiments extended the same logic. Germ-free C9orf72-deficient mice developed little systemic or neural inflammation until bacteria or bacterial communities were introduced. Colonization with P. merdae enhanced inflammation, and gut-directed glycogen digestion promoted survival while reducing microglial reactivity.1
Microglial reactivity means activation-like changes in the brain’s resident immune cells. In ALS/FTD, microglial activation can sit near degenerating neural tissue, but this study points upstream: gut microbial glycogen may help set the peripheral immune tone that reaches the nervous system in a genetically vulnerable host.
Prior C9orf72 Work Already Pointed to Gut-Immune Modulation
Burberry et al. previously showed that gut bacteria could modulate systemic and neural inflammation in C9orf72-deficient mice.3 McCourt et al. make that earlier observation more concrete by naming a molecular class: inflammatory forms of bacterial glycogen with short glucan chains and dense branching.
DeJesus-Hernandez et al. helped establish C9orf72 repeat expansion as a major ALS/FTD cause.2 That genetics-first fact should keep the current study calibrated. Microbial glycogen is not presented as a universal ALS cause. It is a plausible environmental and microbiome amplifier in a C9orf72-linked immune context.
Human ALS microbiome studies have reported fecal community differences, but many have struggled to move from association to function.4 The new study’s strength is functional testing: macrophage cytokines, microbial transcripts, enzyme digestion, germ-free colonization, survival, and human fecal activity were connected in the same argument.
What This Can and Cannot Support
Supported: C9orf72 loss of function can make myeloid cells unusually responsive to specific gut-bacterial glycogen forms; alpha-amylase-sensitive inflammatory glycogen was enriched in small ALS/FTD fecal samples; mouse experiments showed a modifiable gut-to-neuroinflammation pathway.
Not supported: diagnosing ALS from stool, treating ALS with digestive enzymes, or assuming that every ALS case has the same microbiome mechanism. The strongest causal evidence is preclinical, and the human sample was cross-sectional.
Best next test: longitudinal C9orf72-carrier cohorts should measure fecal glycogen activity before symptom onset, during conversion, and during progression. The useful question is whether this microbial product predicts inflammatory transition better than broad microbiome composition alone.
Why the Result Is More Specific Than “Gut Dysbiosis”
Dysbiosis is a broad term for an altered microbial community. It can be useful as a first-pass description, but it often hides the real biological question: which microbial product, sensed by which host pathway, changes which disease-relevant process?
McCourt et al. narrowed that chain by connecting pro-inflammatory fecal environments, glycogen-biosynthesis transcripts, macrophage tumor necrosis factor release, alpha-amylase sensitivity, P. merdae colonization, and microglial reactivity in the same experimental sequence.1
Reader-facing consequence: the practical research target becomes microbial glycogen structure and immune sensing. That distinction keeps the finding useful without turning it into a generic probiotic or microbiome-restoration claim.
Patient Translation Would Need a Carrier-Specific Test
C9orf72-linked ALS/FTD differs from sporadic ALS, SOD1-linked ALS, and other motor-neuron disease categories. The human fecal panel included sporadic ALS, C9orf72-ALS, SOD1-ALS, C9orf72-FTD, and healthy controls, but the sample was too small to define a disease-specific diagnostic threshold.
A serious clinical test would need to separate at least 3 things: whether inflammatory glycogen is common before symptoms, whether it rises near conversion, and whether it predicts progression after diagnosis. It would also need to account for diet, antibiotics, geography, constipation, feeding-tube use, medication exposure, and disease-stage differences that can change the gut microbiome without causing ALS/FTD biology.
Best use now: the paper gives ALS/FTD researchers a functional assay to carry forward. It does not give patients a stool-test answer or a do-it-yourself enzyme treatment.
That conservative boundary is what makes the mechanism credible: it identifies a testable inflammatory product without pretending that a small fecal panel can solve ALS/FTD heterogeneity.
The immediate research value is operational. Future studies can measure glycogen-sensitive cytokine activity, sequence the same fecal communities, and test whether the assay tracks genetic status, disease stage, or inflammatory biomarkers better than broad bacterial taxonomy.
Why C9orf72 status changes the read: C9orf72 is involved in immune-cell handling of intracellular trafficking and inflammatory responses. In this paper, the microbial product did not become important because glycogen is generically toxic. It became important because C9orf72-deficient myeloid cells reacted differently to a bacterial carbohydrate load. That gene-by-product framing is narrower, but it is also more biologically useful.
A broad ALS microbiome claim would be easy to overstate. ALS patients can develop constipation, altered diet, weight loss, medication exposure, respiratory problems, feeding changes, and reduced mobility, all of which can reshape stool samples after disease has already begun.
Functional readouts: McCourt et al. partly avoid that trap by testing macrophage tumor necrosis factor release, alpha-amylase sensitivity, bacterial colonization, and mouse survival. Functional readouts are harder to dismiss than taxonomy alone.
The translation problem: even if inflammatory glycogen is reproducible, a useful marker would need to show timing, specificity, and actionability. The strongest version would appear before rapid decline, concentrate in the genetically vulnerable subgroup, and fall when a disease-relevant outcome improves. A stool assay that only mirrors late-stage gut disruption would be scientifically interesting but clinically weak.
Cautious interpretation: this is one of the cleaner gut-brain mechanisms in ALS/FTD because it names a bacterial product, a host genetic vulnerability, an immune-cell response, and an enzyme-sensitive intervention point.
It remains preclinical and small-sample human evidence, not a patient-ready microbiome protocol.
Questions About C9orf72, Gut Glycogen, and ALS/FTD
Does this mean ALS starts in the gut?
No. The study supports a gut-linked inflammatory amplifier in C9orf72-related biology. ALS remains a nervous-system disease with genetic, cellular, and environmental contributors.
Why is glycogen important here?
Glycogen was the bacterial product that made the immune response experimentally modifiable. Digesting it reduced macrophage cytokine release and improved survival in the mouse model.
Could alpha-amylase become a treatment?
Possibly as a research direction. Current clinical use would be premature because human dosing, targeting, safety, and clinical benefit have not been shown.
Why call this more than a microbiome study?
Because the paper moved past species lists. It identified a microbial chemical feature, tested how C9orf72-deficient immune cells responded, and changed the response enzymatically.
References
- McCourt BT, Limone F, Ventrice D, et al. C9orf72 in myeloid cells prevents an inflammatory response to microbial glycogen. Cell Reports. 2025. doi:10.1016/j.celrep.2025.116906
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011. doi:10.1016/j.neuron.2011.09.011
- Burberry A, Wells MF, Limone F, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 2020. doi:10.1038/s41586-020-2288-7
- Rowin J, Xia Y, Jung B, Sun J. Gut inflammation and dysbiosis in human motor neuron disease. Neurobiology of Aging. 2017. doi:10.1016/j.neurobiolaging.2017.09.007