
In APP/PS1 Alzheimer's model mice, deleting PTP1B or treating with the allosteric inhibitor DPM-1003 improved memory behavior, lowered amyloid burden, and pushed microglia toward SYK-driven clearance — a plausible mechanism, not a human treatment result yet.1
Research Highlights
- PTP1B loss improved behavior in APP/PS1 mice. Genetic deletion and DPM-1003 treatment improved novel object recognition and Morris water maze performance without an obvious motor explanation.1
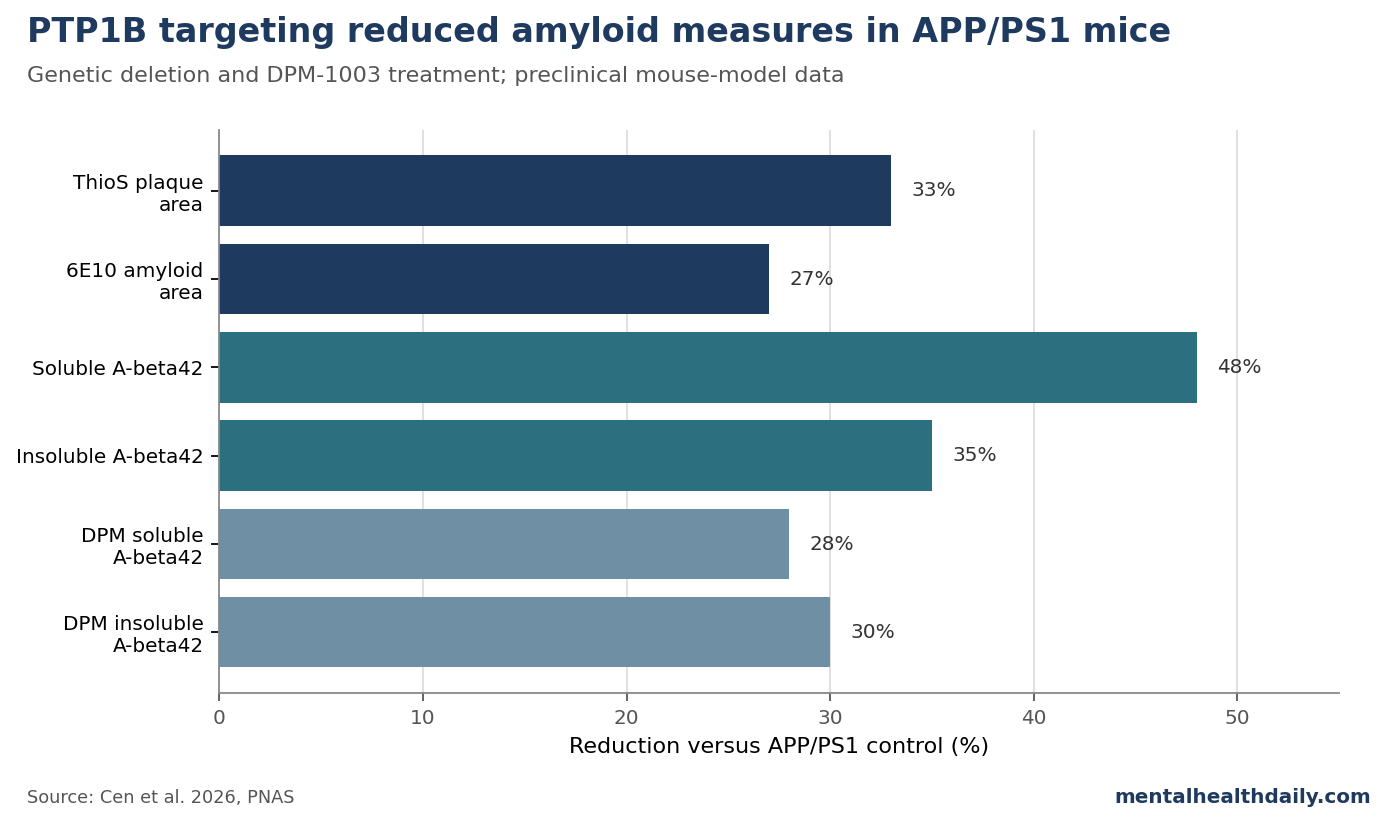
- Amyloid burden fell across several measures. PTP1B deletion reduced ThioS-positive plaque area by 33%, 6E10-positive area by 27%, soluble amyloid-beta 42 by 48%, and insoluble amyloid-beta 42 by 35%.1
- The mechanism was clearance, not less amyloid production. APP, APP C-terminal fragments, BACE1, and presenilin 1 were not changed in a way that explained the amyloid reduction.1
- Microglia looked more active and phagocytic. PTP1B-deficient microglia showed more disease-associated microglia features, more amyloid engulfment, and more CD68-positive phagolysosomal amyloid.1
- SYK is the key link. The authors identified SYK as a direct PTP1B substrate, and SYK inhibition blocked the enhanced phagocytosis and metabolic activation.1
The specific claim is microglial, not a broad anti-amyloid cure: PTP1B may act as a brake on amyloid clearance, and removing that brake may make microglia better able to engulf amyloid in an amyloid-heavy mouse model.
PTP1B Was Tested in APP/PS1 Alzheimer's Model Mice
Cen et al. used the APP/PS1 mouse model, which develops amyloid accumulation beginning around 6 months and cognitive deficits around 12 months. They tested 2 routes: cross-breeding APP/PS1 mice with PTP1B-knockout mice and treating APP/PS1 mice with DPM-1003, an allosteric PTP1B inhibitor.1
A compact study snapshot keeps the result grounded:
- Model: APP/PS1 mice, a widely used amyloid-pathology model of Alzheimer's disease.
- Genetic arm: APP/PS1 mice with or without PTP1B deletion, tested around 12–13 months.
- Drug arm: APP/PS1 mice treated with DPM-1003 for 5 weeks starting at 11 months.
- Behavioral tests: novel object recognition and Morris water maze, with open-field measures used to check locomotor or anxiety-like confounds.
- Mechanism assays: amyloid staining, ELISA, single-cell RNA sequencing, primary microglial phagocytosis assays, Seahorse metabolism, immunoblotting, SYK inhibition, and substrate trapping.1
The behavioral signal was not the only outcome. Both PTP1B deletion and DPM-1003 improved recognition and spatial-memory measures in APP/PS1 mice. The researchers also reported that swimming speed and locomotor measures did not explain the water-maze findings, reducing the chance that motor differences contaminated cognitive interpretation in the mouse studies.1
Amyloid Reductions Pointed Toward Better Clearance
The amyloid data are the centerpiece. In the hippocampus, PTP1B deletion reduced ThioS-positive plaque area by 33% and 6E10-positive area by 27%. DPM-1003 treatment produced similar but slightly smaller reductions: 28% for ThioS-positive area and 26% for 6E10-positive area.1
The biochemical amyloid-beta 42 results were also meaningful. PTP1B deletion reduced soluble amyloid-beta 42 by 48% and insoluble amyloid-beta 42 by 35%. DPM-1003 reduced soluble amyloid-beta 42 by 28% and insoluble amyloid-beta 42 by 30%.1
That still leaves a key question: did PTP1B loss reduce amyloid production, or improve amyloid clearance? The paper leans toward clearance. PTP1B deletion reduced amyloid levels without changing full-length APP, APP C-terminal fragments, BACE1, or presenilin 1 in a way that would explain reduced amyloid production.1
Microglia Shifted Toward a Phagocytic State
Microglia are the brain's resident immune cells. In Alzheimer's disease, they can cluster around plaques, engulf amyloid, secrete inflammatory molecules, and sometimes become metabolically dysfunctional. The therapeutic challenge is that “activate microglia” is too crude. Some activation states may help amyloid clearance; others may worsen inflammation or synaptic injury.
The PTP1B paper found that Ptpn1, the gene encoding PTP1B, was highly expressed in immune cells including microglia. In APP/PS1 mice lacking PTP1B, microglia showed a transcriptional shift toward immune activation and phagocytosis. Disease-associated microglia increased modestly from 44.9% to 51.7%, and upregulated genes included cathepsins, lysosomal proteins, MHC-related genes, Trem2, Axl, Cd9, Csf1, and Spp1.1
The functional assays matched the transcript story. PTP1B-deficient primary microglia had higher baseline phagocytic activity and responded more strongly to amyloid-beta oligomers. In vivo, PTP1B-deficient microglia engulfed more amyloid, and amyloid inside CD68-positive phagolysosomes more than doubled.1
TREM2, SYK, and microglial metabolic fitness have become central to modern Alzheimer's research because amyloid clearance depends on whether microglia have the signaling and energy state required to do the work.4,5
SYK Connected PTP1B to Microglial Energy and Amyloid Uptake
SYK, or spleen tyrosine kinase, is an immune-signaling enzyme. In microglia, SYK sits downstream of several receptors involved in amyloid response and phagocytosis.
SYK mechanism: PTP1B directly dephosphorylates SYK, so deleting or inhibiting PTP1B allows stronger SYK activation, which then supports AKT-mTOR signaling, metabolism, and phagocytosis.1
The energy side is not decorative. Activated microglia need fuel. PTP1B-deficient microglia showed enhanced AKT-mTOR signaling, increased lactate production, higher extracellular acidification rate, increased basal oxygen consumption, increased maximal respiration, and greater mitochondrial-linked ATP production.1
The strongest causal support came from SYK inhibition. BAY61-3606 reduced SYK phosphorylation and brought the enhanced phagocytic activity of PTP1B-deficient microglia down to levels seen in treated wild-type microglia. It also reduced AKT-mTOR signaling and metabolic activation.1
PTP1B Remains a Mouse-Model Target, Not a Human Treatment
The paper should be read with real interest and real restraint. APP/PS1 mice are useful for amyloid biology, but they do not reproduce the full human Alzheimer's disease syndrome. Human disease includes tau, vascular injury, aging immune changes, synaptic vulnerability, comorbid metabolic disease, heterogeneous genetics, and decades-long timing.
The model also starts from an amyloid-heavy genetic background, which makes it well suited for amyloid-clearance biology but cleaner than sporadic late-life Alzheimer's disease. A future test would need to ask whether PTP1B inhibition improves synaptic integrity, neurodegeneration, inflammation balance, and tau-related outcomes alongside plaque and amyloid-beta 42 measures.
There is also a drug-development problem. PTP1B has been considered difficult to target because protein tyrosine phosphatases have conserved, polar catalytic sites. Allosteric inhibitors are one way around that problem, and DPM-1003 is mechanistically interesting, but brain exposure, long-term safety, dose, off-target biology, and human tolerability remain separate questions.1
The amyloid field also carries scars. Anti-amyloid antibodies can reduce amyloid yet produce modest clinical benefits and safety concerns.3 PTP1B inhibition is not the same strategy, because it aims to improve endogenous microglial clearance and metabolic state, but it still has to prove that changing amyloid biology changes human cognition meaningfully.
That is why the most honest interpretation is target validation, not treatment validation. The study says PTP1B is positioned at a biologically important control point: microglial SYK signaling, energy metabolism, and amyloid uptake. It does not yet say that a patient with mild cognitive impairment or Alzheimer's dementia would improve on a PTP1B inhibitor.
How to Interpret PTP1B for Alzheimer's Disease
For mechanism: PTP1B appears to function as a brake on SYK-linked microglial activation in this model. Removing that brake improved amyloid phagocytosis and lowered amyloid burden.
For drug development: the target is plausible, especially because it links metabolism, immune signaling, and amyloid clearance. But DPM-1003 is not an approved Alzheimer's drug, and the current evidence is preclinical.
For amyloid interpretation: this does not rescue every version of the amyloid hypothesis. It supports a narrower claim: amyloid clearance may depend partly on microglial signaling and energy state.
For clinical translation: the next questions are pharmacokinetics, brain penetration, chronic safety, tau and neurodegeneration effects, sex differences, age timing, and whether benefits survive in models beyond APP/PS1.
Evidence-strength note: the study combines mouse genetics, drug treatment, single-cell profiling, and cell assays, so it is stronger than a simple expression association. But it remains preclinical. It can support target biology and pathway plausibility; it cannot establish human dosing, cognitive benefit, long-term immune safety, or whether amyloid clearance through this route changes the clinical course of Alzheimer's disease.
Translation would also need to account for timing. A microglial-clearance strategy may behave differently before plaques accumulate, during mild cognitive impairment, and after widespread tau pathology and synaptic loss. The most useful human program would therefore pair amyloid and tau biomarkers with inflammation markers, cognitive endpoints, and safety monitoring rather than treating plaque reduction alone as the success metric.
Patient selection would be another early challenge. A trial might need amyloid-positive participants with measurable inflammatory or microglial signatures, rather than an all-comers Alzheimer's sample. Without that enrichment, a biologically active PTP1B inhibitor could look weak because many enrolled patients would not have the pathway the drug is meant to change.
Questions About PTP1B and Alzheimer's Models
Why did PTP1B matter in this Alzheimer's mouse study?
PTP1B is protein tyrosine phosphatase 1B, an enzyme that removes phosphate groups from signaling proteins. It is known for roles in insulin, leptin, immune, and metabolic signaling.1
What did PTP1B inhibition do in the Alzheimer's mouse model?
DPM-1003 treatment improved memory behavior and reduced amyloid burden in APP/PS1 mice, including reductions in plaque staining and soluble and insoluble amyloid-beta 42.1
Was SYK necessary for the microglial effect?
The SYK inhibitor BAY61-3606 reduced the enhanced phagocytosis, AKT-mTOR signaling, and metabolic activation seen in PTP1B-deficient microglia.1
Does this mean people should take a PTP1B inhibitor for Alzheimer's disease?
No. The current paper is a mouse and cell-mechanism study. It does not establish human efficacy, dosing, safety, or clinical benefit.
References
- PTP1B Inhibition Promotes Microglial Phagocytosis in Alzheimer's Disease Models by Enhancing SYK Signaling. Cen Y, Alves SR, Song D, et al. Proceedings of the National Academy of Sciences. 2026;123(6):e2521944123. doi:10.1073/pnas.2521944123
- The Amyloid Hypothesis of Alzheimer's Disease at 25 Years. Selkoe DJ, Hardy J. EMBO Molecular Medicine. 2016;8:595–608. PubMed
- Evaluation of Clinical Benefits of Treatments for Alzheimer's Disease. Liu KY, et al. Lancet Healthy Longevity. 2023;4:e645–e651. PubMed
- SYK Coordinates Neuroprotective Microglial Responses in Neurodegenerative Disease. Ennerfelt H, et al. Cell. 2022;185:4135–4152.e22. PubMed
- TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Ulland TK, et al. Cell. 2017;170:649–663.e13. PubMed