“Depression is inflammation” is one of the most popularized clinical-neuroscience claims of the last decade. Anti-inflammatory diets, anti-inflammatory supplements, and biomarker-guided depression treatment all sell on the strength of this framing. The data underneath it support a narrower version of the claim than the popular framing suggests: the association is robust in clinical samples, considerably weaker in community samples, and the causal interpretation requires a subtype hypothesis that wasn’t part of the original story.
Research Highlights
- Inflammatory Depression is an accurate framing for a subgroup, not for everyone. Meta-analyses of clinical depression samples consistently find elevated inflammatory cytokines (IL-6, TNF-α, CRP). Studies in community samples with proper confound control find much weaker or null associations — the strong clinical signal does not generalize cleanly.
- A new community study finds essentially no inflammation-depression link. Bruellman et al. 2026, a pre-registered twin study of 972 healthy adults, found neither C-reactive protein (CRP) nor a pro-inflammatory cytokine index was associated with depression symptoms. AUD-cytokine associations that emerged in the full sample disappeared in co-twin control analyses — familial confounders, not direct effects.
- Anti-inflammatory drugs produce a modest pooled effect on depression, but heterogeneity is severe. Meta-analyses of NSAIDs, statins, omega-3, minocycline, and other anti-inflammatory agents in depression find SMDs around −0.55 (Kohler-Forsberg 2019), but with high publication bias and effects driven by a few small trials. The clinical translation hasn’t replicated cleanly.
- An “inflammatory subtype” of depression is the most defensible reading. Inflammation likely plays a causal role for some depressed patients — particularly those with elevated baseline CRP, atypical features, or comorbid metabolic dysfunction — without being the underlying mechanism for depression broadly. Stratified trials (e.g., Raison’s infliximab study) support this picture.
- The inflammation-depression literature is the strongest contemporary case study in why clinical-sample findings need community-sample replication. Effects that look causal in patient cohorts may reflect reverse causation, confounding by health-behavior, or selection into clinical care. Don’t read clinical-sample results as universal biology.
What the Clinical-Sample Literature Found
The contemporary inflammation-depression literature traces to two foundational meta-analyses.
Howren and colleagues’ 2009 meta-analysis in Psychosomatic Medicine pooled CRP (C-reactive protein, a general marker of body-wide inflammation) and IL-6 (interleukin-6, an inflammatory signaling protein) data across 41 community and clinical studies. Depressed individuals had small but consistent elevations — on the order of 0.15 to 0.25 standard deviations above non-depressed controls.1
Dowlati and colleagues’ 2010 meta-analysis in Biological Psychiatry focused on patients with clinical depression and reported larger elevations in IL-6 and TNF-α (tumor necrosis factor alpha, another core inflammatory signaling protein) specifically.2
Subsequent meta-analyses (Liu et al. 2012; Köhler et al. 2018) extended these findings across additional cytokines (cell-signaling molecules the immune system uses to coordinate the inflammatory response), generally confirming elevated IL-6, TNF-α, IL-1β, and CRP in patients with major depressive disorder relative to healthy controls.3
Miller and Raison’s 2016 review in Nature Reviews Immunology synthesized this work into the “inflammation hypothesis of depression.” In plain terms: peripheral inflammation (in the body) signals into the brain through several routes, and ends up disturbing the same circuits depression already involves — the brain’s stress-hormone system (HPA axis), the chemistry of mood-regulating neurotransmitters (monoamines like serotonin and dopamine), and the reward and motivation networks that go quiet in depression.4
Three pieces of supporting evidence are routinely cited:
- Interferon-induced depression. Patients receiving interferon-alpha for hepatitis C develop major depressive symptoms at high rates (around 30 to 40%), establishing that experimentally induced peripheral inflammation can produce clinically significant depression.5
- Endotoxin challenge studies. Healthy volunteers given low-dose lipopolysaccharide (LPS) develop transient anhedonia, fatigue, and depressed mood within hours, with stronger symptoms in participants with higher baseline CRP.6
- Anti-inflammatory treatment trials. Meta-analyses of randomized trials of NSAIDs, statins, omega-3 fatty acids, minocycline, and cytokine antagonists report pooled SMDs (standardized mean differences, which let us compare studies that used different symptom scales) around −0.55 versus placebo on depression symptom scales.7
The clinical-sample case is reasonably strong. The community-sample case is much weaker.
Why Community Samples Find a Smaller (or Absent) Signal
Several large community-sample studies have failed to replicate the clinical-sample inflammation-depression association after proper confound control.
Figueroa-Hall and colleagues’ 2022 study in Translational Psychiatry analyzed 1,724 community adults and found that the apparent CRP-depression association was entirely explained by confounders — primarily BMI, sleep, and exercise.8 When these were properly modeled, the residual CRP-depression effect was indistinguishable from zero.
A new community-sample study by Bruellman and colleagues, published in 2026 in Brain, Behavior, & Immunity Health, sharpens this picture.9 The team analyzed 972 community adults (mean age 33; range 28 to 49), including a subset of same-sex twin pairs from the Colorado Adoption/Twin Study of Lifespan behavioral development. Two pre-registered hypotheses were tested:
- Higher cytokine levels would be associated with more depression symptoms.
- Heavier alcohol use and AUD symptoms would be associated with higher cytokine levels.
Neither was supported. The headline findings:
- MDD symptoms were not associated with the pro-inflammatory index (composite of IL-1β, IL-6, and TNF-α) or with CRP in either unadjusted or covariate-adjusted models.9
- AUD symptoms were associated with lower circulating cytokines (IL-1β, IL-4, IL-10, IL-12) after Bonferroni correction — in the opposite direction from the prediction.9
- Co-twin control analyses, which compare twin pairs to each other and remove genetic and shared-environment confounding, eliminated the inverse AUD-cytokine associations. Within twin pairs, the twin with higher AUD did not have proportionally lower cytokines. The full-sample associations reflected familial confounding, not direct effects of alcohol on cytokines.9
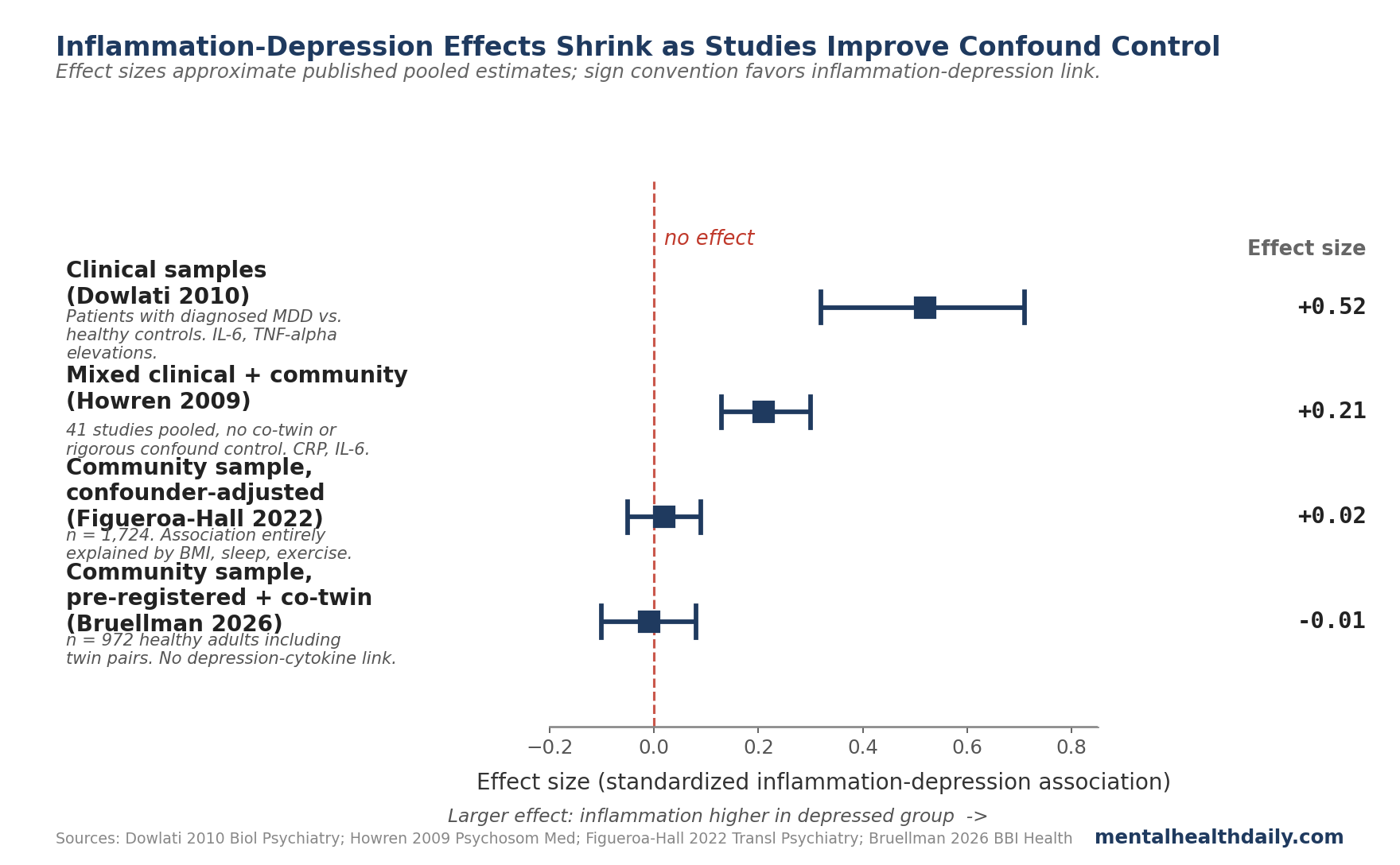
The pattern across studies is informative. Clinical samples (severely depressed patients in residential or specialty care) show the strongest cytokine elevations. Community samples without confound control show smaller effects. Community samples with proper confound control or co-twin design show effects close to zero.
The straight reading: the inflammation signal in clinical-sample studies mostly reflects how sick those patients are overall — severe depression itself, the medical illnesses that travel with it, the medications they’re on, and the way sicker people end up at specialty clinics in the first place. It’s not the underlying biology of depression in the general population.
Anti-Inflammatory Treatment Trials: A More Modest Signal Than Headlines Suggest
If inflammation causes depression, anti-inflammatory drugs should treat depression. The trial literature is mixed but more positive than the community-sample observational data alone would predict.
Köhler-Forsberg and colleagues’ 2019 meta-analysis in Acta Psychiatrica Scandinavica pooled 36 RCTs of anti-inflammatory pharmacological agents across heterogeneous patient populations.7 Pooled SMD versus placebo was approximately −0.55 — a moderate-to-large effect, comparable to standard antidepressant trials. The meta-analysis covered NSAIDs, statins, omega-3 fatty acids, glucocorticoids, minocycline, modafinil, N-acetylcysteine, and cytokine inhibitors.
Three caveats deserve weight:
- The trials disagree wildly on how big the effect is. The heterogeneity score (I²) exceeds 70% across most drug classes — meaning the studies don’t agree on the magnitude. The pooled number averages across drugs that work in completely different ways: common painkillers, omega-3 supplements, and monoclonal antibody injections that block specific inflammatory proteins. The patients also vary in how inflamed they were to begin with. The aggregate signal points the right way, but you can’t pin down the exact size from this kind of mixed-bag pool.
- Publication bias is detectable. Funnel plot asymmetry in Köhler-Forsberg’s analysis suggests small flattering trials are over-represented in the literature. The trim-and-fill adjusted effect estimate (which simulates the missing null trials) shrinks meaningfully but remains nonzero.
- Trials that split patients by their starting inflammation tell the clearest story. Raison and colleagues’ 2013 RCT tested infliximab (an injected drug originally developed for rheumatoid arthritis that blocks TNF-α signaling) versus placebo in treatment-resistant depression. There was no benefit overall — but a meaningful benefit in the patients whose starting CRP was already high (above 5 mg/L, a signal of ongoing low-grade inflammation).10 This is the strongest empirical support for the “inflammatory subtype” idea: anti-inflammatory treatment helps the people whose depression actually has an inflammatory signature, not depression generally.
The Inflammatory-Subtype Hypothesis Is What the Data Suggest
The most defensible synthesis: a clinically meaningful subgroup of depressed patients have inflammatory signatures, respond to anti-inflammatory treatment, and may have biology that fits the inflammation hypothesis — while the broader depressed population does not.
Markers proposed to identify this subgroup include:
- Elevated baseline CRP (typically > 3 mg/L, sometimes > 5 mg/L). The infliximab trial used this as a stratifier.10
- Atypical depression features (hypersomnia, hyperphagia, leaden paralysis, mood reactivity), which correlate with inflammation more reliably than melancholic features.
- Comorbid metabolic dysfunction (obesity, insulin resistance, metabolic syndrome). The “immune-metabolic” subtype proposed by Penninx and colleagues integrates these.
- Treatment resistance to monoaminergic antidepressants, particularly when accompanied by inflammatory comorbidity.
Miller’s 2025 review explicitly frames this as a precision-psychiatry application: not “depression is inflammation” but “for some depressed patients, inflammation is part of the picture, and stratification matters.”11 The evidence base for this narrower claim is genuinely better than for the broader version.
Where the Inflammation-Depression Data Are Weakest
Several limitations cut across the inflammation-depression literature.
Cross-sectional dominance limits causal inference. Most observational studies measure cytokines and depression symptoms at a single timepoint. The forward causal inference (inflammation → depression) and the reverse (depression → inflammation, via behavioral pathways like sedentariness and disrupted sleep) are both consistent with the data.
Health behaviors muddy the picture. Depressed people exercise less, sleep worse, eat differently, and smoke more — all of which independently raise inflammatory markers on their own. Even careful statistical adjustment can’t fully strip those effects out, so some of what looks like a direct depression-inflammation link is probably just behavior.
Cytokine measurement is noisy and timing-sensitive. Single blood draws capture cytokines at one moment; cytokines fluctuate substantially across the day, post-prandial state, and recent infections. Studies with single timepoint measurements have lower signal-to-noise than studies using repeated sampling.
Publication bias appears in both directions. Positive associations were over-published in early decades; null community-sample findings have been historically harder to publish. The Bruellman 2026 pre-registered design helps with this, but most of the existing literature wasn’t pre-registered.
Clinical-to-community generalization is the central methodological gap. The strong clinical-sample literature was built largely on inpatients and specialty-clinic samples. Generalizing to the broader depressed population was mostly assumed, not demonstrated.
Practical Conclusions From the Calibrated Inflammation Evidence
The current state of the literature supports a few clear practical conclusions and rules out a few popular ones.
- Don’t take “depression is inflammation” as a universal mechanism. The framing fits a subgroup of depressed patients reasonably well and the broader depressed population poorly. Acting as if it explains depression generally overshoots what the evidence supports.
- Don’t take cytokine panels for depression diagnosis. Commercial inflammation-panel-based depression products lack the predictive validity their marketing implies. CRP alone has weak discriminative value at the individual level even in studies that find statistical group-level associations.
- Anti-inflammatory diet and exercise have other health benefits worth pursuing on their own merits. If a Mediterranean-pattern diet, regular exercise, and adequate sleep also happen to lower inflammatory markers and modestly improve mood, that’s a fine secondary benefit. They’re worth doing for cardiovascular and metabolic reasons regardless.
- For treatment-resistant depression with elevated CRP, anti-inflammatory augmentation has the strongest evidence. Raison’s infliximab data and subsequent stratified trials suggest patients with treatment-resistant depression and CRP > 3 mg/L are reasonable candidates for trials of cytokine-targeted augmentation in research settings. This is not yet first-line care.
- Address health-behavior confounders before reaching for biomarker-guided treatment. BMI, sleep quality, physical activity, and diet quality account for much of the cytokine-depression association in community samples. Addressing those is good standard care and likely captures most of the practically relevant inflammation-mediated path.
Reader Questions on the Inflammation-Depression Link
Is depression caused by inflammation?
For a subgroup of depressed patients, plausibly yes — particularly those with elevated baseline CRP, atypical features, or metabolic comorbidity. For the broader depressed population, the case is much weaker. Clinical-sample meta-analyses find consistent cytokine elevations2; community samples with proper confound control find weaker or null effects.8,9
Should I get a CRP test to find out if I have inflammatory depression?
Not as a routine step. CRP is useful as a stratifier in research trials but has weak individual-level predictive validity for depression treatment response. If you’re already getting routine bloodwork that includes high-sensitivity CRP for other reasons (cardiovascular risk assessment), the result is informative; ordering it specifically for depression workup is premature.
Do anti-inflammatory drugs treat depression?
Modestly, on average, and unevenly across drug classes. Köhler-Forsberg 2019’s meta-analysis found a pooled effect of about −0.55 standard deviations versus placebo, but the trials disagreed wildly on the size and there’s clear publication bias (small flattering trials are over-represented).7 The strongest evidence (Raison 2013, infliximab) suggests the benefit is concentrated in patients who started with high inflammation, not depression generally.10
Will an anti-inflammatory diet help my depression?
Possibly, with caveats. Mediterranean-pattern diets show small benefits on depression in trials (SMILES, HELFIMED), though the mechanism may be nutritional rather than specifically anti-inflammatory. The dietary pattern has clear cardiovascular and metabolic benefits regardless of mood effect, so it’s a reasonable thing to do on broader grounds.
Why do clinical-sample studies show such large inflammation effects when community samples don’t?
Three reasons:
- Clinical samples have more severe depression, which itself drives more behavioral inflammation (sedentary, poor sleep, poor diet).
- Clinical samples have more comorbid medical illness, which independently elevates inflammation.
- Clinical samples have more medication exposure, including SSRIs and other agents that interact with cytokine signaling. Together these confounders inflate the apparent direct depression-cytokine association.8,9
What about omega-3s for depression?
EPA-predominant omega-3 supplementation shows small-to-moderate benefits in depression meta-analyses, with stronger effects at doses > 1 g/day EPA and in patients with major depressive disorder rather than subclinical symptoms. The benefit may or may not be inflammatory in mechanism — omega-3s have many effects on neural lipid composition independent of cytokine modulation.
References
- Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Howren MB, Lamkin DM, Suls J. Psychosomatic Medicine. 2009;71(2):171-186. doi:10.1097/PSY.0b013e3181907c1b
- A meta-analysis of cytokines in major depression. Dowlati Y et al. Biological Psychiatry. 2010;67(5):446-457. doi:10.1016/j.biopsych.2009.09.033
- Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Kohler CA et al. Acta Psychiatrica Scandinavica. 2018;135(5):373-387. doi:10.1111/acps.12698
- The role of inflammation in depression: from evolutionary imperative to modern treatment target. Miller AH, Raison CL. Nature Reviews Immunology. 2016;16(1):22-34. doi:10.1038/nri.2015.5
- Neuropsychiatric symptoms associated with hepatitis C and interferon alpha: a review. Dieperink E, Willenbring M, Ho SB. American Journal of Psychiatry. 2000;157(6):867-876. doi:10.1176/appi.ajp.157.6.867
- Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Eisenberger NI et al. Biological Psychiatry. 2010;68(8):748-754. doi:10.1016/j.biopsych.2010.06.010
- Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Kohler-Forsberg O et al. Acta Psychiatrica Scandinavica. 2019;139(5):404-419. doi:10.1111/acps.13016
- Psychiatric symptoms are not associated with circulating CRP concentrations after controlling for medical, social, and demographic factors. Figueroa-Hall LK et al. Translational Psychiatry. 2022;12(1):279. doi:10.1038/s41398-022-02049-y
- Inflammation, mental health, and alcohol behaviors: Testing links leveraging a familial community sample. Bruellman R et al. Brain, Behavior, & Immunity – Health. 2026;53:101229. doi:10.1016/j.bbih.2026.101229
- A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression. Raison CL et al. JAMA Psychiatry. 2013;70(1):31-41. doi:10.1001/2013.jamapsychiatry.4
- Beyond inflammation: the immune-metabolic basis of depression. Miller AH. Trends in Cognitive Sciences. 2025;29(2):109-122. doi:10.1016/j.tics.2024.10.001