
A 2026 neuron and mouse study found that CB1 receptors accumulate when autophagic flux is blocked, but p62 deficiency did not significantly increase steady-state CB1 receptor abundance in brain or hypothalamus and did not make CB1 signaling the primary explanation for late-onset obesity.1 The result separates 2 claims that are easy to blur: CB1 can be processed through autophagy, while p62-related obesity can still arise through other metabolic pathways.
Research Highlights
- Autophagy affected CB1: blocking autophagic flux with Bafilomycin A1 produced substantial CB1 receptor accumulation in primary cortical neurons.1
- p62 did not raise steady CB1: p62 knockout mice showed no significant CB1 protein-abundance increase in whole brain or hypothalamus.1
- Obesity still appeared: p62 knockout mice developed late-onset obesity without hyperphagia, plus early hypoactivity and elevated hypothalamic 2-AG with age.1
- CB1 antagonism did not solve it: pharmacological CB1 receptor antagonism failed to uncover a direct CB1-dependent mechanism for the phenotype.1
- Human translation is limited: the study used 0 clinical participants, and prior CB1 antagonist trials already showed psychiatric risk in humans.2
CB1 receptors are cannabinoid receptors widely expressed in the brain, where they regulate synaptic release, appetite, reward, stress, pain, and energy balance. Autophagy is a cellular recycling system that moves damaged proteins and organelles toward lysosomal degradation.
p62, also called sequestosome 1, is an autophagy receptor that helps tag cargo for degradation. Because p62 knockout mice become obese with age, Keller et al. tested whether disrupted CB1 receptor handling could explain the metabolic phenotype.
That question sits at the crossing of 3 literatures that often get collapsed into a single story: CB1 metabolism papers, p62-autophagy obesity papers, and receptor-trafficking work. The cleaner read is narrower. CB1 signaling remains metabolically powerful, p62 loss clearly produces an obesity phenotype in several mouse models, and Keller et al. add a receptor-turnover result; the study does not show that p62 obesity is mainly a CB1 receptor-abundance disorder.
CB1 Receptors Accumulated When Autophagy Was Blocked
In primary cortical neurons from wild-type mice, Bafilomycin A1 inhibited autophagic flux and produced substantial CB1 receptor accumulation. That result supports a basic cell-biology claim: CB1 receptors can be degraded through an autophagy-dependent route.1
Turnover claim: receptor abundance at any moment reflects production, signaling, internalization, recycling, and degradation. Blocking degradation can reveal a pathway even when the same pathway is not the dominant driver of a whole-animal phenotype.
Keller et al. used primary cortical neurons from mouse embryos at embryonic day 15.5–16.5, treated cells for 10 h, and compared Bafilomycin A1 with CB1-active compounds. Bafilomycin A1 is a lysosomal inhibitor: it blocks the late degradation step, so proteins that normally move through the autophagy-lysosome route can accumulate.
CB1 agonist stimulation with HU-210 partly reduced the receptor accumulation caused by autophagy blockade, suggesting that receptor activation can influence turnover. The antagonist SR141716A, better known as rimonabant in the obesity-drug literature, had only a weak antagonistic effect in this cell setup.
Trafficking interpretation: Keller et al. read that drug pattern cautiously. CB1 internalization, recycling, endosomal degradation, and autophagic processing may all contribute, and the experiment was not designed to map every trafficking branch.
The result strengthens the claim that CB1 receptors can enter an autophagy-linked degradation pathway, but it does not turn autophagy into the only CB1 disposal route. Broader receptor-trafficking work has already shown that G-protein-coupled receptors can be sorted toward endolysosomal destruction through multiple ubiquitin-linked pathways.5
A clean MHD reading is therefore: Keller et al. add CB1 to the autophagy-substrate conversation, while leaving receptor-pool location and cell-type specificity unresolved.
p62 Knockout Obesity Was Not Primarily CB1-Driven
The mouse data changed the interpretation. p62 deficiency did not significantly alter CB1 receptor protein abundance in brain or hypothalamus. Hypothalamic ERK1/2 signaling downstream of CB1 was only modestly attenuated, and CB1 receptor antagonism did not reveal a direct receptor-dependent explanation for obesity.1
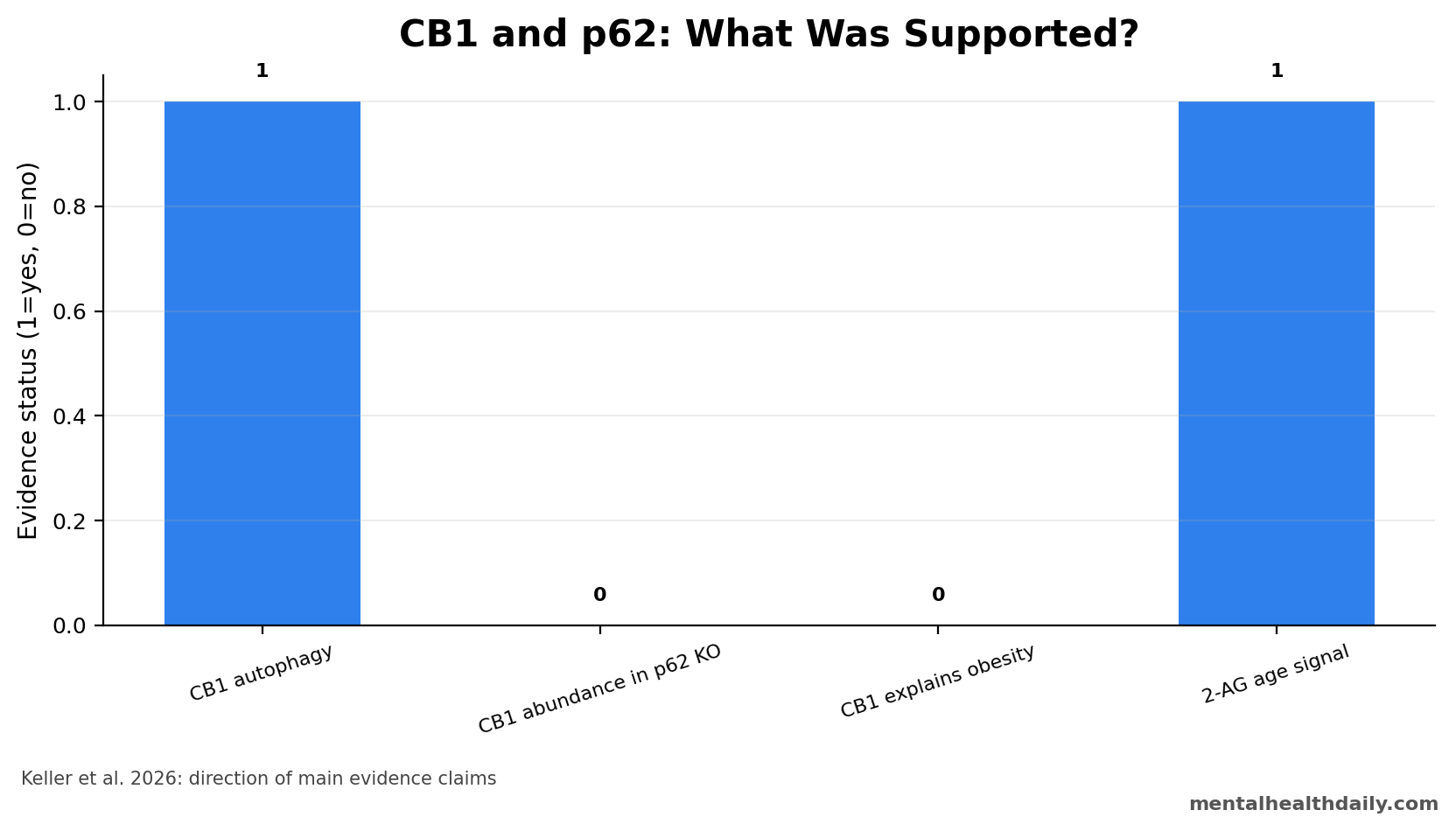
That matters for interpretation because p62 knockout mice still showed a phenotype: late-onset obesity without hyperphagia, early hypoactivity, elevated hypothalamic 2-arachidonoylglycerol (2-AG) with age, and altered fasting-refeeding behavior.
The weight data were directionally straightforward. At 12 weeks, male wild-type and p62 knockout mice were almost identical in body weight: 31.2 g vs. 31.0 g.
By 18 weeks, p62 knockout mice were significantly heavier, at 32.9 g vs. 28.3 g, and by 21 weeks the genotype gap exceeded 6 g. Food intake did not rise enough to explain that divergence, which argues against a simple “ate more, gained more” interpretation.1
The activity data supplied the more plausible behavioral bridge. Reduced voluntary locomotion appeared around 15 weeks, roughly 3 weeks before the significant weight separation.
In 1-year-old males, p62 knockout mice weighed 18 g more than controls and were active for about 1.7 min per h during the active phase, compared with 3.6 min per h in wild-type mice. That does not prove hypoactivity is the full cause of obesity, but it is much closer to the phenotype than steady-state CB1 receptor abundance.
2-AG is an endogenous cannabinoid, meaning a lipid signaling molecule made by the body that can activate cannabinoid receptors. Elevated hypothalamic 2-AG keeps the endocannabinoid system in the picture, even though the primary driver did not appear to be simple CB1 receptor accumulation.
Tissue specificity also matters. Keller et al. found elevated 2-AG in hypothalamus, while anandamide (AEA) and arachidonic acid (AA) were unchanged and white-adipose-tissue and liver endocannabinoid measures did not show the same broad genotype difference. That pattern fits a central signaling hypothesis better than a body-wide endocannabinoid excess claim.
p62 Obesity Has a Broader Autophagy and Thermogenesis Backstory
The CB1-negative result is easier to understand against prior p62 work. A 2006 Cell Metabolism study reported mature-onset obesity and insulin resistance in p62-deficient mice, placing p62 upstream of metabolic regulation before Keller et al. asked the CB1-specific question.6 Later work linked p62 to beta-adrenergic input, mitochondrial function, and thermogenesis, which are more direct routes into energy expenditure than receptor abundance alone.7
A separate 2021 adipocyte paper pushed the interpretation further: NBR1, another selective autophagy receptor, became a key piece in repressing thermogenesis in p62-deficient adipocytes through PPARγ signaling.8 NBR1 means neighbor of BRCA1 gene 1, a selective autophagy receptor that can overlap with p62-like cargo-handling roles. PPARγ is a nuclear receptor that helps regulate adipocyte differentiation and lipid storage.
Those adjacent papers make the CB1 result less surprising. If p62 deficiency can disturb adipocyte thermogenesis, mitochondrial function, and selective autophagy compensation, then normal bulk CB1 abundance is not a paradox. The phenotype can stay real while the first suspected receptor route fails.
The practical synthesis is not “CB1 is irrelevant.” It is more specific: CB1 biology plausibly modifies appetite, locomotion, and lipid storage, but Keller et al. did not find the clean molecular bridge from p62 loss to CB1 receptor accumulation in the hypothalamus or whole brain.
CB1 Biology Is Relevant to Metabolism, but the Human Drug History Is Cautionary
The endocannabinoid system remains a credible metabolic target. CB1 activation can increase food intake, alter reward-related eating, and influence energy expenditure. Human pharmacology already tested this logic: rimonabant, a CB1 antagonist, reduced weight and improved some metabolic outcomes, but psychiatric adverse effects helped end its use.2
The older mechanistic literature explains why rimonabant looked attractive. Cota et al. showed that endogenous cannabinoid signaling affected energy balance through central orexigenic drive and peripheral lipogenesis, linking brain appetite circuits with fat-storage biology.9 Later reviews treated the endocannabinoid system as a major organizer of obesity and metabolic disease, not a minor pathway.10
But drug translation exposed the penalty for a centrally acting CB1 strategy. In the RIO-Europe trial, rimonabant produced weight and cardiometabolic improvements in overweight or obese patients, yet the broader program became clinically unacceptable because depression, anxiety, and other psychiatric adverse events were not side details for a brain-active appetite drug.2
Mechanistic bottom line: Keller et al. refine the mechanism rather than revive a simple drug idea. Their data support CB1 autophagic turnover in neurons, while arguing against a direct CB1-centered explanation for p62 knockout obesity.
That distinction is especially important for cannabis-adjacent interpretation: a mouse autophagy result does not become advice about cannabis, CB1 blockade, or human weight loss.
What the 2026 Mouse Study Can and Cannot Support
The strongest supported claim is cellular: CB1 receptor protein accumulated when autophagic degradation was blocked in primary cortical neurons. The strongest negative claim is whole-animal: p62 deficiency did not significantly increase bulk CB1 receptor abundance in brain or hypothalamus, and CB1 antagonism did not uncover a direct receptor-dependent obesity mechanism.
Several constraints keep the conclusion narrow:
- Bulk tissue can miss compartments: whole-brain and hypothalamic lysates do not separate surface receptors, internalized receptors, autophagosomal pools, or neuron-specific subtypes.
- Mouse age changes the phenotype: juvenile p62 knockout mice did not show the same fasting-refeeding pattern as adult and aged animals, so timing is part of the biology.
- Endocannabinoids are not receptors: elevated hypothalamic 2-AG suggests altered signaling tone, but it is not equivalent to increased CB1 receptor abundance or increased CB1 causality.
- Human inference is indirect: 0 clinical participants were studied, and the relevant human CB1 antagonist history is limited by psychiatric tolerability.
Evidence-strength note: this was a mechanistic mouse and cell study. It can clarify receptor biology and p62 phenotype interpretation. It cannot tell cannabis users how CB1 affects weight, predict psychiatric effects of CB1 drugs, or define human obesity treatment.
Questions About CB1 Autophagy and p62 Obesity
Did p62 deficiency increase CB1 receptor levels?
No significant steady-state increase was found in brain or hypothalamus, even though CB1 receptors accumulated when autophagic flux was blocked in neurons.
Why can both statements be true?
A receptor can use autophagy for turnover while whole-animal obesity is driven by other pathways. Cellular degradation routes and organism-level metabolic phenotypes do not map one-to-one.
Does this change cannabis-obesity advice?
No. The study is about CB1 turnover and p62 knockout mice, not cannabis exposure, appetite behavior, or human weight management.
Why does the rimonabant history matter here?
It keeps the interpretation grounded. CB1 blockade can affect weight biology, but central CB1 drugs have carried psychiatric risk in humans, so a mouse receptor-turnover result should not be read as a near-term obesity-treatment signal.
References
- Keller C, Rading S, Candur B, Bindila L, Loers G, Karsak M. Exploring cannabinoid receptor CB1 autophagy and the obesity phenotype of p62-deficient mice. Biochemistry and Biophysics Reports. 2026;46:102571. https://doi.org/10.1016/j.bbrep.2026.102571
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients. Lancet. 2005. https://doi.org/10.1016/s0140-6736(05)66374-x
- Lu HC, Mackie K. An introduction to the endogenous cannabinoid system. Biological Psychiatry. 2016. https://doi.org/10.1016/j.biopsych.2015.07.028
- Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007. https://doi.org/10.1016/j.cell.2007.10.035
- Dores MR, Trejo J. Endo-lysosomal sorting of G-protein-coupled receptors by ubiquitin: diverse pathways for G-protein-coupled receptor destruction and beyond. Traffic. 2019;20:101–109. https://doi.org/10.1111/tra.12619
- Rodriguez A, Duran A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metabolism. 2006;3:211–222. https://doi.org/10.1016/j.cmet.2006.01.011
- Muller TD, Lee SJ, Jastroch M, et al. p62 links beta-adrenergic input to mitochondrial function and thermogenesis. Journal of Clinical Investigation. 2013;123:469–478. https://doi.org/10.1172/jci64209
- Huang J, Linares JF, Duran A, et al. NBR1 is a critical step in the repression of thermogenesis of p62-deficient adipocytes through PPARgamma. Nature Communications. 2021;12:2876. https://doi.org/10.1038/s41467-021-23085-0
- Cota D, Marsicano G, Tschop M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. Journal of Clinical Investigation. 2003;112:423–431. https://doi.org/10.1172/jci17725
- Mazier W, Saucisse N, Gatta-Cherifi B, Cota D. The endocannabinoid system: pivotal orchestrator of obesity and metabolic disease. Trends in Endocrinology & Metabolism. 2015;26:524–537. https://doi.org/10.1016/j.tem.2015.07.007