
A 2025 Science Advances mouse study found a more brain-dependent metformin mechanism than the usual liver-gut framing suggests: low-dose metformin lost its glucose-lowering effect when brain Rap1 signaling could not be reduced, and 36 of 47 ventromedial-hypothalamus SF1 neurons responded directly to metformin.1
Research Highlights
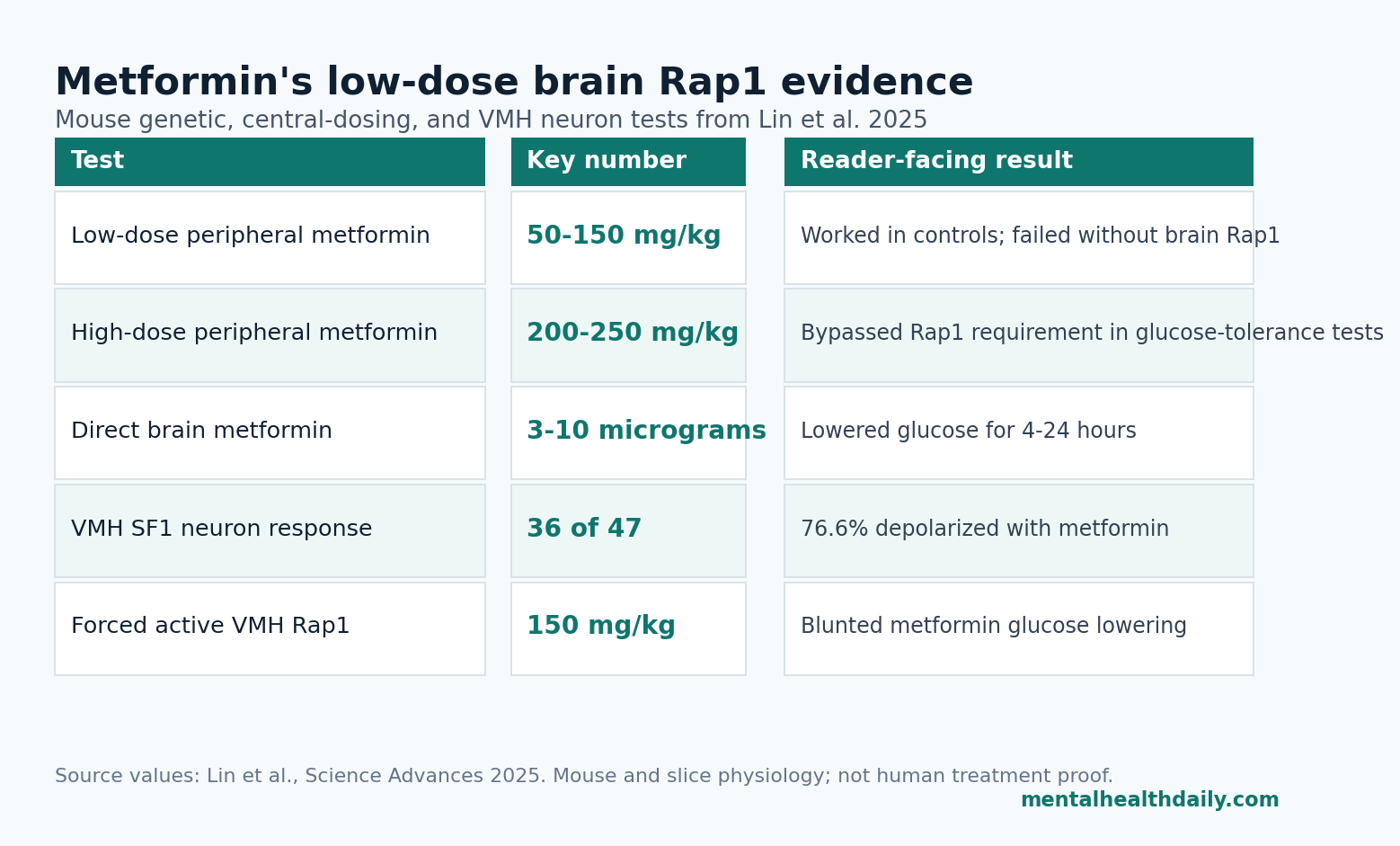
- Low-dose metformin needed brain Rap1: Control mice lowered glucose after 50 to 150 mg/kg metformin, while forebrain Rap1 knockout mice lost the low-dose effect and recovered responsiveness only at higher 200 to 250 mg/kg doses.1
- Central dosing was active: Direct brain delivery of 3 to 10 micrograms metformin reduced glucose for 4 to 24 hours, and 1 microgram activated c-Fos in the ventromedial hypothalamus rather than nearby metabolic nuclei.1
- VMH SF1 neurons responded directly: Metformin depolarized 36 of 47 SF1 neurons (76.6%) at 1 to 100 micromolar, and Rap1 deletion largely abolished that neuronal response.1
- Dose translation is the key caveat: The paper framed 10 to 40 micromolar blood concentrations as therapeutically relevant in humans, while high experimental doses may bypass the brain Rap1 requirement through other pathways.1,2
- The mechanism adds rather than replaces: Lysosomal AMPK, hepatic glucose production, gut signaling, and mitochondrial effects still remain part of metformin biology; the Rap1 study adds a brain-control layer, not a one-pathway explanation.2,3
Metformin is a biguanide drug used for type 2 diabetes and diabetes prevention, mainly because it lowers glucose with low cost and a long safety record. The classic explanation is peripheral: less hepatic glucose output, altered gut signaling, mitochondrial effects, and changes in cellular energy sensing.2
Rap1 is a small guanosine triphosphatase, meaning a molecular switch that toggles signaling pathways on and off. Lin et al. focused on Rap1 in the brain because prior work had already shown that hypothalamic Rap1 can change whole-body glucose balance, including liver glucose output, skeletal-muscle glucose uptake, and insulin sensitivity.4
50 to 150 mg/kg Metformin Failed When Brain Rap1 Was Deleted
Lin et al. used high-fat-diet mice with forebrain-specific Rap1 deletion and compared metformin against several other glucose-lowering drugs. The distinction was specific: metformin failed to significantly lower glucose in Rap1ΔCNS mice, while rosiglitazone, exendin-4, glibenclamide, dapagliflozin, and insulin still lowered glucose in a broadly normal way.1
Why the comparator drugs matter: if Rap1 deletion simply made the animals unable to lower glucose, every drug would have looked weak. Instead, the deficit clustered around metformin. That pattern makes the result more informative than a single knockout-versus-control contrast.
Low-dose split: metformin at 50, 100, and 150 mg/kg reduced glucose in control mice but not in Rap1ΔCNS mice.
High-dose bypass: during glucose-tolerance testing, 50 and 100 mg/kg lost effect in Rap1ΔCNS mice, while 200 and 250 mg/kg restored glucose-lowering ability.1
That dose split is central to the paper’s claim. Low-dose metformin was the condition where brain Rap1 looked necessary. Higher doses could apparently use Rap1-independent routes, which fits the broader metformin literature: different concentrations can recruit different cellular mechanisms.2
Direct Brain Metformin Lowered Glucose at 3 to 10 Micrograms
The researchers then asked whether brain exposure could directly drive the effect. Intracerebroventricular dosing means injecting a compound into the brain’s ventricular fluid space so researchers can test central nervous system effects more directly than ordinary peripheral dosing allows.
Metformin delivered into the brain at 3 to 10 micrograms reduced blood glucose for 4 to 24 hours in diet-induced obese mice. The effect also appeared in DIO/STZ mice, a model combining diet-induced obesity with streptozotocin-related beta-cell injury, and in ob/ob mice, a leptin-deficient obesity model.1
Rap1 still controlled the central effect: direct brain metformin did not further lower glucose in brain-specific Rap1-deficient mice. The paper also reported that central metformin reduced hypothalamic Rap1 activity, placing Rap1 downstream of metformin exposure rather than merely beside it.
A central pathway can lower glucose at small brain doses and becomes difficult to ignore when low-dose peripheral metformin also fails in mice missing brain Rap1. Ordinary clinical metformin may still recruit liver, gut, mitochondrial, and lysosomal mechanisms at the same time.
VMH SF1 Neurons Were the Brain Node, Not a Generic Brain Effect
Ventromedial hypothalamic nucleus (VMH) refers to a hypothalamic region involved in energy balance, glucose regulation, defensive behavior, and autonomic output. SF1 neurons are VMH neurons marked by steroidogenic factor 1, a transcription factor used here to target a defined glucose-control cell population.
After central metformin, c-Fos staining increased in the VMH at 1 microgram. c-Fos is a common marker of recent neuronal activation. Nearby metabolic regions, including the dorsomedial hypothalamus and arcuate nucleus, did not show the same activation pattern.1
Electrophysiology made the VMH finding more direct. In hypothalamic slices, metformin at 1 to 100 micromolar depolarized 36 of 47 SF1 neurons (76.6%) and modestly increased firing. When Rap1 was deleted from SF1 neurons, the metformin-induced depolarization and firing response were largely abolished.1
Locking Rap1 Active Blunted Metformin’s Effect
Loss-of-function evidence can be hard to interpret because deleting a signaling molecule can create developmental compensation or baseline differences. Lin et al. therefore also used gain-of-function tests: they forced Rap1 into an active state, then asked whether metformin still worked.
Forebrain expression of constitutively active Rap1 raised fasting glucose and impaired glucose tolerance in high-fat-diet mice. More importantly, metformin at 150 mg/kg improved glucose tolerance in controls but not in Rap1CNSV12 mice.1
VMH-targeted viral delivery tightened the location. Expressing active Rap1 in the VMH attenuated metformin’s glucose-lowering effect during acute and chronic experiments and weakened its ability to improve glucose tolerance. The simplest interpretation is that metformin needs to reduce Rap1 activity in VMH SF1 neurons; if Rap1 is genetically locked active, the drug cannot push the pathway in the same direction.
Rap1 Adds a Brain Layer to Metformin Biology
Metformin has never had one clean mechanism. LaMoia and Shulman emphasized that concentration matters because low, clinically relevant exposures and high experimental exposures can trigger different biology.2 Ma et al. later identified a low-dose lysosomal AMPK pathway involving PEN2 and v-ATPase, showing that low-dose metformin can act through specific molecular machinery rather than vague cellular stress.3
Rap1 fits that pattern. Lin et al. measured 22 micromolar metformin in serum and 4.6 micromolar in hypothalamus after 150 mg/kg peripheral dosing, putting hypothalamic exposure in the range where VMH SF1 neurons responded in slices. The paper also cited prior work showing cerebrospinal-fluid and brain metformin levels around 0.5 to 10 micromolar after clinically relevant circulating exposure.1
Calibrated read: the brain Rap1 pathway is strongest as a low-dose neural control mechanism, not as a replacement for hepatic, gut, mitochondrial, or lysosomal models. Higher metformin doses bypassed the Rap1 requirement, so a one-mechanism headline would misread the dose biology.
Animal Evidence Does Not Prove Human Brain Benefits
Evidence-strength note: this was a mouse and slice-physiology study. It can show that manipulating brain Rap1 changes metformin’s glucose effect in mice. It cannot prove that human patients require the same pathway, that Rap1 explains individual metformin response, or that metformin should be used for cognition, mood, aging, or psychiatric symptoms.
Baseline glucose also complicates the knockout interpretation. The researchers acknowledged that Rap1-deficient mice sometimes had lower baseline glycemia, which can create a floor effect: if glucose is already lower, there is less room for metformin to reduce it. They argued against a simple floor-effect explanation by pointing to glycemia-matched cohorts and retained responsiveness to other drugs, but the limitation remains relevant.1
A human test would need to separate central exposure from ordinary peripheral response, ideally by pairing metformin pharmacokinetics with glucose outcomes and a Rap1-pathway marker.
It would also need to show whether hypothalamic Rap1 adds predictive value beyond dose, kidney function, gut tolerance, insulin resistance, and baseline glucose.
Human brain-relevant metformin research exists, but it is not the same question. Gantois et al. reported metformin benefits in a fragile X mouse model, and Koenig et al. tested metformin in a small Alzheimer disease crossover pilot.5,6 Those papers help establish that metformin can matter in neurobiological contexts, but they do not validate Rap1 as a human clinical target.
Questions About Metformin and Brain Rap1
Does this mean metformin works through the brain in people?
Not proven. The study makes a strong mouse case that low-dose metformin can require brain Rap1 signaling, but human confirmation would need pharmacokinetic, genetic, imaging, or biomarker evidence tied to actual glucose outcomes.
Does this make metformin a mental-health drug?
No. The measured endpoint was glucose control. Brain involvement does not automatically mean psychiatric benefit, cognitive enhancement, or anti-aging efficacy.
Why does low dose matter so much?
Low-dose experiments were designed to approximate clinically relevant exposure. High experimental concentrations may recruit pathways that never dominate during ordinary treatment, so the low-dose Rap1 result is more pharmacologically meaningful than a high-dose-only mechanism would be.
What would make the Rap1 pathway clinically useful?
The useful next step would be a human-response marker: evidence that metformin reaches hypothalamic targets at relevant concentrations, that Rap1-pathway variation predicts glucose response, or that a brain-facing intervention changes metformin sensitivity without unsafe central effects.
References
- Lin H-Y, Lu W, He Y, et al. Low-dose metformin requires brain Rap1 for its antidiabetic action. Science Advances. 2025;11:eadu3700. doi:10.1126/sciadv.adu3700
- LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocrine Reviews. 2021;42:77-96. doi:10.1210/endrev/bnaa023
- Ma T, Tian X, Zhang B, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. 2022;603:159-165. doi:10.1038/s41586-022-04431-8
- Kaneko K, Lin H-Y, Fu Y, et al. Rap1 in the VMH regulates glucose homeostasis. JCI Insight. 2021;6:e142545. doi:10.1172/jci.insight.142545
- Gantois I, Khoutorsky A, Popic J, et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nature Medicine. 2017;23:674-677. doi:10.1038/nm.4335
- Koenig AM, Mechanic-Hamilton D, Xie SX, et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Disease and Associated Disorders. 2017;31:107-113. PubMed