A 2026 Nature Communications study found that Mecp2 overexpression deregulated approximately 5,000 genes in mouse neural progenitor cells but only approximately 500 mostly small-change genes in mature neurons, with the same progenitor-vs-neuron split reproduced in human iPSC-derived cells.1 For Rett syndrome gene therapy, that is the useful calibration: extra MeCP2 is most dangerous when it reaches developmentally plastic progenitor cells, not automatically whenever mature neurons receive more protein.
Research Highlights
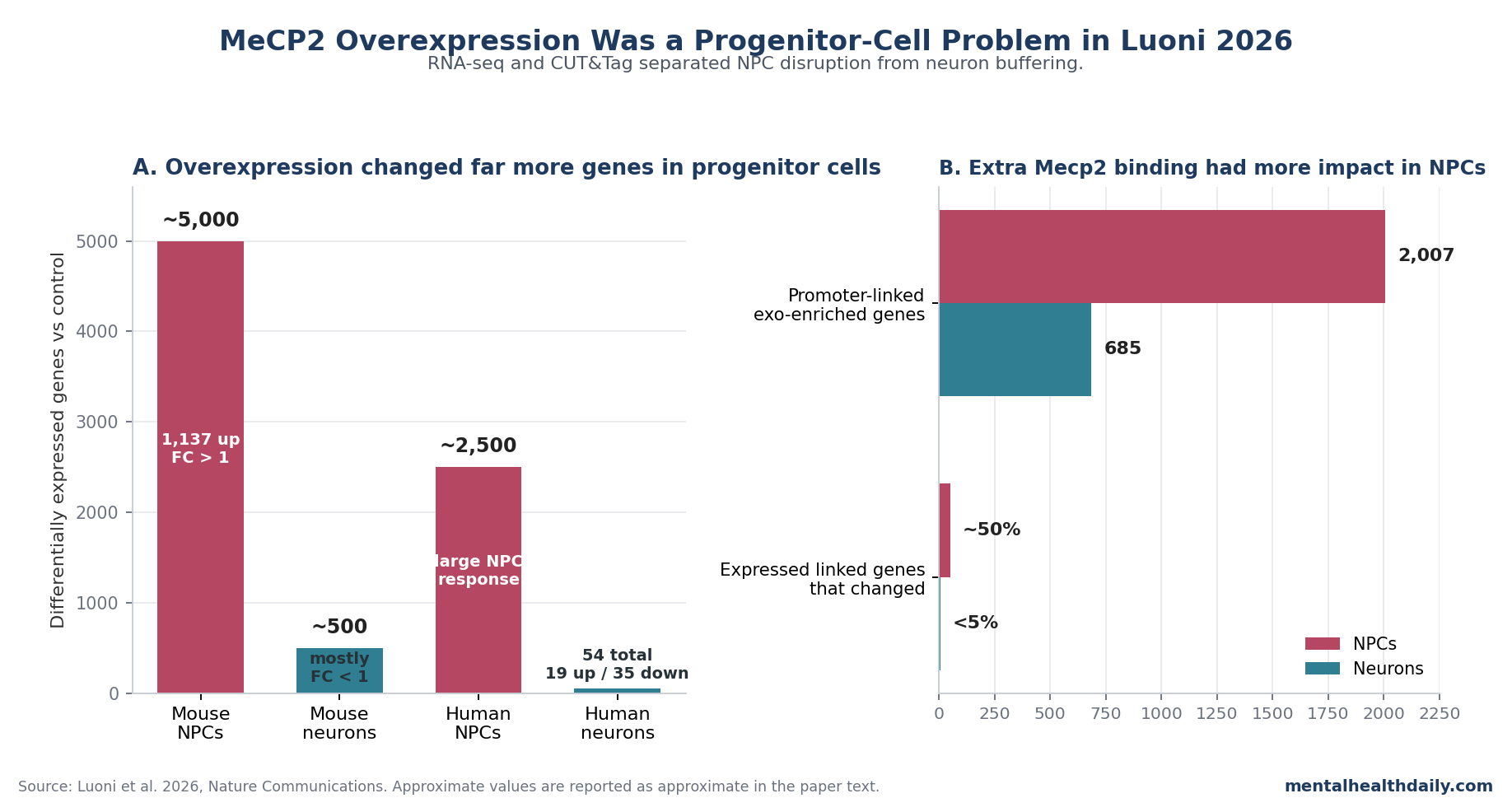
- Progenitor cells carried the headline effect: mouse neural progenitor cells showed approximately 5,000 differentially expressed genes after Mecp2 overexpression, including 1,137 upregulated genes with fold change >1; mature neurons showed approximately 500 deregulated genes, mostly with fold change <1.1
- Human cells repeated the same split: human iPSC-derived neural progenitor cells showed approximately 2,500 deregulated genes, while human neurons showed only 19 upregulated and 35 downregulated genes, all below fold change 1 except MECP2 itself.1
- Binding was shared, but impact was not: endogenous and exogenous Mecp2 targeted roughly 15,000 CpG islands with >95% co-occupancy, yet exogenous enrichment mapped to 2,007 promoter-linked genes in progenitors vs 685 in neurons.1
- Bivalent genes explain the developmental risk: over 1/3 of NPC exogenous-enriched genes had bivalent chromatin, about 40% of bivalent NPC genes were not yet expressed, and excess Mecp2 recruited SWI/SNF machinery toward activation of cell-fate genes.1
- Mature neurons had 2 buffering mechanisms: many CpG islands were already occupied by endogenous Mecp2, and exogenous Mecp2 degraded about 50% faster than endogenous Mecp2 in neurons after cycloheximide exposure, with significant differences at 4 h and 8 h.1
MeCP2 is a methyl-CpG-binding protein: it reads DNA methylation and chromatin state, then helps tune gene expression in the nervous system. Too little functional MeCP2 causes Rett syndrome, usually through loss-of-function mutations in MECP2; too much MeCP2 causes MECP2 duplication syndrome, a severe neurodevelopmental disorder marked by hypotonia, developmental delay, recurrent infections, seizures, and progressive neurological problems.2,3
That 2-sided dosage biology is why Rett gene therapy has always carried an uncomfortable question. AAV-based therapies try to deliver working MECP2 to the brain, but many cells in females with Rett syndrome already express the healthy X-linked allele.
The fear is concrete: chronic developmental overexpression of Mecp2 causes neurological disease in mice.9 Luoni et al. narrowed the question from “is more MeCP2 toxic?” to “which cells, at which developmental stage, can actually absorb the extra dose?”1
5,000 Progenitor-Cell DEGs vs About 500 Mostly Small Neuron Changes
Luoni et al. overexpressed Mecp2 in primary mouse neural progenitor cells (NPCs; dividing cells that can still choose neural fates) and mature neurons (post-mitotic cells already committed to neuronal identity). Both systems achieved roughly 3- to 4-fold Mecp2 overexpression over baseline, with significant protein increases in NPCs (p = 0.044) and neurons (p = 0.013).1
The RNA-seq result was not close. NPCs showed approximately 5,000 differentially expressed genes (DEGs), and 1,137 of those were upregulated with fold change >1.
Mature neurons showed approximately 500 DEGs, but the researchers emphasized that most changes had fold change <1, meaning the neuron response was numerically detectable but biologically much smaller.
That direction also cut against the old shorthand that MeCP2 is simply a transcriptional repressor. In NPCs, excess Mecp2 drove more strong upregulation than strong downregulation, and gene-ontology analysis pointed toward developmental and neural-differentiation programs.
Excess Mecp2 was pushing immature cells toward inappropriate activation of fate-control genes.
Human validation made the result harder to dismiss as a mouse-culture artifact. Human iPSC-derived NPCs showed approximately 2,500 DEGs after MECP2 overexpression.
Human neurons showed 19 upregulated and 35 downregulated genes, and all of those changes had fold change <1 except MECP2 itself.1
Extra Mecp2 Targeted Similar CpG Islands but Hit Progenitor Cells Harder
The binding data explain why the same protein increase can land so differently. CpG islands are DNA regions rich in cytosine-guanine sequence sites, often near gene promoters where transcription is regulated.
Luoni et al. found that endogenous and exogenous Mecp2 targeted roughly 15,000 CpG islands, with more than 95% of those sites occupied by both forms of the protein across NPCs and neurons.1
The shared target map did not mean equal biological effect. In neurons, endogenous Mecp2 is already abundant, so many CpG islands are already covered before the transgene arrives.
In NPCs, endogenous Mecp2 is low, leaving more open promoter-associated territory for exogenous protein to occupy.
Using an enrichment-score threshold, the researchers identified 2,287 CpG islands enriched for exogenous Mecp2 in NPCs; 89% were promoter localized, yielding 2,007 linked genes. Neurons had fewer exogenous-enriched promoter-linked targets, with 685 associated genes and 81.5% promoter localization.1
The transcriptional consequence was even more asymmetric. Around 75% of the linked genes were expressed in both cell types, but nearly 50% of expressed NPC-linked genes were deregulated vs fewer than 5% in neurons.
Within the NPC exogenous-enriched gene list, upregulated genes roughly doubled downregulated genes, and strong upregulation was more common: 203 of 449 upregulated genes had fold change >1, compared with 57 of 288 downregulated genes.1
Bivalent Developmental Genes Were the Vulnerable Substrate
Bivalent genes are developmental genes held in a poised state: their promoters carry both activating H3K4me3 marks and repressive H3K27me3 marks, so the gene is not fully on or off. That poised state is useful in progenitor cells because it keeps fate decisions available until the right developmental signal arrives.7
Luoni et al. found that over 1/3 of the Mecp2-exogenous-enriched genes in NPCs were bivalent. About 40% of the bivalent Mecp2-enriched genes in NPCs were not yet expressed, making them plausible targets for inappropriate activation.
In neurons, only 16% of exogenous-enriched genes were bivalent, and 83% of those were already transcriptionally active.1
That is a clean developmental mechanism. NPCs contain silent or low-expression fate genes waiting for a commitment signal.
Excess Mecp2 lands on CpG-rich promoters, recruits activation machinery, and shifts some of those genes toward premature expression. Mature neurons have fewer silent bivalent targets left to flip.
The study team then connected this activation to SWI/SNF, a chromatin-remodeling complex that helps open DNA and activate developmental programs. Co-immunoprecipitation supported a physical association between MeCP2 and SMARCB1, a SWI/SNF subunit.
CUT&Tag showed Smarcb1 enrichment at Mecp2 target genes in NPCs, especially bivalent genes such as Zic3, Grin3a, and Reln, which were upregulated in the RNA-seq data.1
Chromatin-state data pointed in the same direction without overstating the effect. 4f-SAMMY-seq, a method that separates more open euchromatin from more compact heterochromatin, found almost no chromatin-state shifts in neurons (S2S-to-S3 = 0%; S3-to-S2S = 0.04%).
NPCs showed more heterochromatin-to-euchromatin movement (S3-to-S2S = 0.42%), and the affected regions included around 300 genes enriched for cell-cycle transition and neuronal differentiation.1
Mature Neurons Buffered Extra Mecp2 Through Saturation and Faster Turnover
Two neuron-specific defenses explain why mature cells did not respond like progenitors.
Binding-site saturation: endogenous Mecp2 already occupied many neuronal CpG island targets. When exogenous Mecp2 arrived, it did not create a large new binding program because the preferred sites were already filled.
Salt-extraction assays supported weaker chromatin association for exogenous Mecp2 in neurons: the exogenous protein was almost entirely released in the 400 mM NaCl fraction, while endogenous Mecp2 showed a broader distribution including the 600 mM fraction.1
Protein turnover: cycloheximide experiments tested how fast the protein decayed after new protein synthesis was blocked. In NPCs, endogenous and exogenous Mecp2 had similar stability.
In neurons, exogenous Mecp2 degraded approximately 50% faster than endogenous Mecp2, with significant differences after 4 h for FLAG-tagged Mecp2 (p = 0.027) and after 8 h for V5- and FLAG-tagged exogenous Mecp2 (p = 0.005).1
Mature neurons could still respond to larger doses, longer exposure, or different vectors. At the studied overexpression range and time scale, they had 2 buffers that progenitor cells lacked: the chromatin sites were already crowded, and extra protein was less stable.
Embryonic Overexpression and Adult AAV Exposure Did Not Behave the Same
Rett gene therapy ultimately depends on intact-brain biology as well as cell-culture assays. Luoni et al. used E14.5 in utero electroporation to overexpress Mecp2 during embryonic cortical development, then compared that with adult brain overexpression using AAV-PHP.eB delivered systemically at 1*10^11 vector genomes per mouse.1
Embryonic exposure: Mecp2 overexpression increased the migration of EGFP-positive cells into the cortical plate and accelerated neuronal differentiation, matching the NPC result: when excess Mecp2 reaches progenitor-stage cells, it can push developmental timing.
Adult exposure: AAV-mediated Mecp2 overexpression did not produce the same electrophysiological disruption. Patch-clamp recordings found that embryonic NPC exposure increased spontaneous excitatory postsynaptic current frequency and amplitude in adult brain tissue, while adult AAV:Mecp2 exposure did not differ from AAV:EGFP control.1
That split fits older MHD-relevant evidence better than a simple “too much MeCP2 is toxic” rule. Collins et al. showed that chronic Mecp2 overexpression beginning early in life causes progressive neurological disease in mice.9
Guy et al. showed that postnatal Mecp2 restoration can reverse neurological defects in a Rett mouse model.8
Sinnett et al. reported improved survival after MECP2 gene therapy in MeCP2-null mice without apparent neurological toxicity from overexpression after intracisternal delivery.4 Luoni et al. add a mechanism: embryonic progenitor exposure and mature-neuron exposure are different biological experiments.
What This Does and Does Not Prove for Rett Gene Therapy
For Rett syndrome, the finding is reassuring in a specific way. It supports the idea that postnatal delivery to mature neurons may have a wider safety margin than MECP2 duplication syndrome would imply from the outside.
Mature neurons can buffer extra protein through CpG island saturation and proteostasis, and the adult AAV mouse experiment did not reproduce the embryonic overexpression phenotype.1
Clinical safety still has to come from TSHA-102, NGN-401, or other human trial programs. Clinical AAV products differ in promoter design, dose, distribution, immune response, cell-type tropism, and duration.
Females with Rett syndrome are also cellular mosaics: some cells express the mutant X chromosome, while others express the wild-type copy and already have functional MeCP2. The relevant clinical problem is cell-by-cell dose control, not average dose in a dish.
Several limits should stay visible:
- Wild-type model systems: most mechanistic assays used wild-type cells rather than Rett-mutant mosaic tissue.
- Vector mismatch: lentiviral overexpression in cultured cells is not the same as clinical AAV delivery in children.
- Cell-type coverage: the study focused on NPCs and neurons; astrocytes, oligodendrocytes, peripheral tissues, and adult neurogenic niches may not follow the same rules.
- Time horizon: the experiments clarify acute and medium-term biology, while clinical gene therapy needs years-long safety data.
The best reading is calibrated, not dismissive. MECP2 duplication syndrome remains the warning sign for developmental overexpression.
Luoni et al. show why postnatal neuronal delivery can diverge from that warning sign without pretending that every dose, vector, or cell target is safe.
Reader Questions on MeCP2 Dose in Rett Gene Therapy
Why would neural progenitor cells be more vulnerable than mature neurons?
Neural progenitor cells have low endogenous MeCP2 and still carry many poised developmental genes. Extra Mecp2 can occupy promoter CpG islands and help activate fate-control genes through SWI/SNF machinery.
Mature neurons already express high MeCP2, have many occupied CpG island targets, and degrade exogenous protein faster.1
Does the 5,000-vs-500 DEG split mean neurons had no response?
No. The neuron RNA-seq analysis detected approximately 500 deregulated genes, but most had fold change <1, and human neurons had only 54 deregulated genes with fold change <1 except MECP2.
The better wording is “buffered,” not “unchanged.”1
How does this affect the duplication-syndrome concern?
MECP2 duplication syndrome still shows that excess MeCP2 can be harmful, especially across development. The Luoni study narrows the risk model: embryonic or progenitor-cell overexpression is biologically different from postnatal mature-neuron exposure, so duplication syndrome should not be treated as a simple one-to-one prediction for every Rett gene-therapy dose.3,9
What safety signal would be most mechanism-specific in future studies?
A mechanism-specific safety readout would look for inappropriate activation of bivalent developmental genes, especially in progenitor-like or neurogenic cell populations. General adverse-event tracking remains necessary, but this paper points to SWI/SNF-linked activation of cell-fate genes as the specific molecular hazard to monitor.1
Should families read this as proof that Rett gene therapy is safe?
No. This is mechanistic preclinical evidence, not clinical proof.
It explains why mature-neuron delivery may be safer than the duplication-syndrome analogy suggests, but phase 1/2 and later trial data remain the evidence that can establish human safety, dose tolerance, and durability.5,6
References
- Luoni M, Kubacki M, Giannelli SG, et al. MeCP2 gene dosage-dependent neurodevelopmentally restricted defects arise by aberrant activation of cell fate-determining bivalent genes. Nature Communications. 2026;17:3225. doi:10.1038/s41467-026-71432-w
- Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics. 1999;23(2):185–188. doi:10.1038/13810
- Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. American Journal of Medical Genetics Part A. 2010;152A(5):1079–1088. doi:10.1002/ajmg.a.33184
- Sinnett SE, Hector RD, Gadalla KKE, et al. Improved MECP2 gene therapy extends the survival of MeCP2-null mice without apparent toxicity after intracisternal delivery. Molecular Therapy: Methods & Clinical Development. 2017;5:106–115. doi:10.1016/j.omtm.2017.04.006
- ClinicalTrials.gov. A Phase 1/2/3 Study of TSHA-102 Gene Therapy in Females With Rett Syndrome (REVEAL Pivotal Study). ClinicalTrials.gov identifier: NCT05606614. ClinicalTrials.gov
- ClinicalTrials.gov. A Novel, Regulated Gene Therapy (NGN-401) Study for Females With Rett Syndrome (Embolden). ClinicalTrials.gov identifier: NCT05898620. ClinicalTrials.gov
- Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi:10.1016/j.cell.2006.02.041
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143–1147. doi:10.1126/science.1138389
- Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human Molecular Genetics. 2004;13(21):2679–2689. doi:10.1093/hmg/ddh282
- Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi:10.1126/science.1153252