
A 2026 review of 52 studies found that chronic primary pain is linked to several physiological stress markers, especially cortisol, heart rate, heart-rate recovery, and mean arterial pressure. The pattern is real enough to study, but not clean enough to use as a simple biomarker panel for pain severity.1
Research Highlights
- 52 studies were reviewed: Vyverman et al. synthesized physiological stress markers and experimental pain outcomes across 2,657 people with chronic primary pain or controls.1
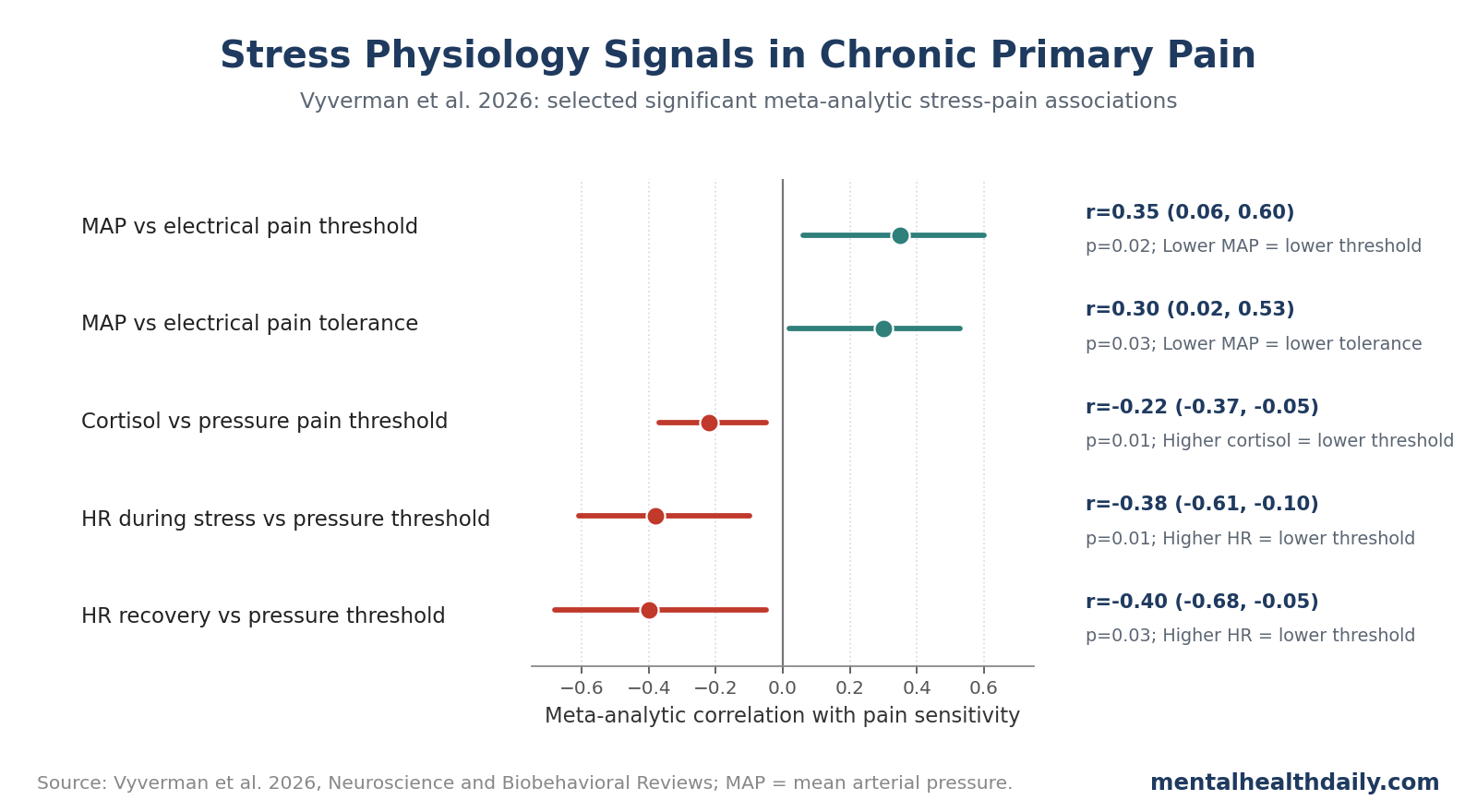
- Cortisol showed a pain-sensitivity signal: higher baseline cortisol correlated with lower pressure pain threshold (r = −0.22, 95% CI −0.37 to −0.05, p = 0.01).1
- Heart-rate findings clustered around stress response: higher heart rate during stress or recovery correlated with lower pressure pain thresholds, with selected pooled r values from −0.38 to −0.53.1
- Mean arterial pressure moved in the opposite direction: higher MAP correlated with higher electrical pain threshold (r = 0.35) and tolerance (r = 0.30), consistent with blood-pressure-linked pain modulation.1
- GRADE certainty varied across 4 levels: evidence certainty ranged from very low to moderate, so the result supports mechanisms and research design more than immediate clinical testing.1
Chronic primary pain refers to persistent pain that cannot be fully explained by tissue damage or another disease process. Fibromyalgia, chronic widespread pain, irritable bowel syndrome pain, and some chronic localized pain syndromes often sit in this category.
52 Studies Tested Stress Physiology Against Pain Sensitivity
Evidence base: Vyverman et al. reviewed 5 cross-sectional studies and 47 case-control studies. The total population was 2,657 participants, including chronic primary pain groups and controls. The review examined the autonomic nervous system, the hypothalamic-pituitary-adrenal (HPA) axis, and experimental pain outcomes.1
The autonomic nervous system controls rapid body-state changes such as heart rate, blood pressure, sweating, and heart-rate variability. Heart-rate variability (HRV) is beat-to-beat variation in heart rhythm; higher vagally mediated HRV often reflects better flexible regulation, though interpretation depends on context.
The HPA axis is the body's hormonal stress-response system. Cortisol is its most familiar output. In pain research, cortisol is not a “stress hormone” in a cartoon sense; it is a timed endocrine signal that changes with sleep, illness, inflammation, trauma exposure, medications, and pain itself.
Timing mattered: The review separated stress timing into baseline, reactivity during a stressor, recovery after a stressor, changes in stress, changes in pain, and combined stress-pain change. A resting marker can miss the main problem if the abnormality appears only during challenge or during recovery.
Pain measurement: The pain outcomes were mostly quantitative sensory testing (QST), a set of lab methods that measures pain threshold, pain tolerance, and pain modulation using standardized stimuli. QST does not capture all real-world pain, but it gives researchers a controlled way to compare pain sensitivity across studies.
Cortisol, Heart Rate, and MAP Were the Clearest Signals
The review reported many non-significant associations, but several meta-analytic findings stood out. The clearest cluster involved higher stress activation paired with greater pain sensitivity.
Selected significant associations included:
- Baseline cortisol vs. pressure pain threshold: r = −0.22, 95% CI −0.37 to −0.05, p = 0.01.
- Heart rate during a stressor vs. baseline pressure pain threshold: r = −0.38, 95% CI −0.61 to −0.10, p = 0.01.
- Heart rate during recovery vs. baseline pressure pain threshold: r = −0.40, 95% CI −0.68 to −0.05, p = 0.03.
- Heart rate during recovery vs. pressure pain threshold during a stressor: r = −0.53, 95% CI −0.71 to −0.27, p < 0.005.
- Heart rate during recovery vs. pressure pain threshold after a stressor: r = −0.48, 95% CI −0.68 to −0.22, p < 0.005.
In these rows, negative correlations mean that higher stress physiology was associated with lower pain thresholds. Lower threshold means pain is reported at a lower stimulus intensity, so it is a higher-sensitivity pattern.
Mean arterial pressure (MAP), the average pressure across a cardiac cycle, showed a different but interpretable signal. Higher MAP correlated with higher electrical pain threshold (r = 0.35, 95% CI 0.06-0.60, p = 0.02) and higher electrical pain tolerance (r = 0.30, 95% CI 0.02-0.53, p = 0.03). In plain English, lower MAP tracked greater sensitivity to electrical pain.1
Autonomic Regulation Fits the Chronic Pain Model
Autonomic findings make biological sense because chronic pain changes more than sensory input. It changes sleep, threat monitoring, movement, avoidance, inflammation, interoception, and stress responsivity.
Several mechanisms can connect autonomic stress markers to pain sensitivity:
- Descending pain modulation: brainstem and cortical systems can dampen or amplify incoming pain signals. Autonomic state may reflect how well those systems regulate pain.
- Baroreflex-linked analgesia: blood-pressure regulation can interact with pain inhibition. Lower MAP signals may mark weaker pressure-linked analgesic control.
- Sympathetic arousal: higher heart rate during stress may reflect a body state primed for threat, which can lower pain thresholds.
- Recovery failure: heart rate that stays elevated after stress may indicate slower return to baseline, which may matter more than the peak response itself.
Calling chronic pain “just stress” is both biologically sloppy and clinically harmful. The better model is shared circuitry: stress-response systems and pain-processing systems can reinforce each other.
That shared circuitry is also why purely structural explanations often disappoint. Imaging, inflammation, autonomic function, endocrine timing, and psychological threat learning may each explain part of chronic primary pain, but no single layer is the whole condition.2,3
HRV Results Were Less Clean Than the Popular Story
Heart-rate variability is often described as a resilience marker, and low HRV is frequently linked to stress-related illness. In this review, HRV did not produce a simple universal finding.
Several baseline HRV contrasts were non-significant, including high-frequency HRV vs. pressure pain threshold, cold pain threshold, and cold pain tolerance. Low-frequency HRV and LF/HF ratio results were also mostly non-significant or context-dependent.1
The mixed pattern is plausible. HRV can shift with respiration, posture, recording duration, medication, sleep debt, anxiety, pain anticipation, and fitness. A 5-minute resting HRV sample may not capture the same biology as HRV during a cold pressor task or after recovery from a social stressor.
That does not make HRV irrelevant. It means HRV may be too dependent on measurement conditions, pain phenotype, medication use, posture, respiration, time of day, and stressor type to function as a standalone chronic-pain marker.
The clinical calibration is direct:
- HRV biofeedback and relaxation training may help some pain patients.
- A single HRV number should not be treated as a pain-severity score.
- Recovery dynamics after stress may be more informative than resting HRV alone.
- Future studies need standardized HRV recording and better pain-phenotype separation.
Stress-Pain Research Needs Better Timing and Phenotyping
The review's biggest contribution is a map of where study design needs to improve.
Chronic primary pain is heterogeneous. A patient with fibromyalgia, widespread pressure sensitivity, trauma history, sleep disruption, and orthostatic symptoms may not share the same stress-pain profile as someone with chronic localized pelvic pain or temporomandibular pain.
Future studies should separate:
- Pain phenotype: widespread vs. localized pain, pressure vs. heat/cold/electrical sensitivity, and presence of central sensitization features.
- Stress phase: baseline, active stressor, immediate recovery, and delayed recovery.
- Stress system: autonomic markers, cortisol, inflammatory markers, and subjective stress should not be collapsed into one “stress” bucket.
- Medication and comorbidity: antidepressants, beta blockers, sleep disorders, nicotine, caffeine, obesity, menstrual-cycle phase, and hormonal therapy can alter stress physiology.
That level of design is harder, but it is the only way to know whether stress markers predict pain trajectories, treatment response, or relapse after improvement.
Intervention studies are especially important. If a treatment improves sleep, reduces catastrophizing, increases movement tolerance, or improves autonomic recovery, researchers should test whether the physiological marker changes alongside pain sensitivity. That is stronger evidence than showing a cross-sectional correlation once.
Examples include exercise programs, cognitive behavioral therapy for pain, HRV biofeedback, sleep treatment, trauma-focused care, and multidisciplinary rehabilitation. None should be sold as a universal fix, but each can test whether shifting stress physiology changes pain processing in a defined subgroup.
Limitations of the Stress-Pain Evidence
- Mostly case-control studies. Case-control designs can show group differences and correlations, but they are weak for causal direction.
- Small meta-analytic cells. Several pooled estimates were based on only 2-5 studies, limiting precision.
- Measurement inconsistency. Studies differed in stressors, timing, QST methods, HRV processing, and cortisol collection.
- Participant imbalance. Many studies enrolled only women, and some did not report sex distribution or age clearly.
- GRADE certainty varied. Evidence certainty ranged from very low to moderate, so clinical translation remains premature.
Questions About Stress Markers and Chronic Pain
Can cortisol diagnose chronic pain?
No. Higher baseline cortisol correlated with lower pressure pain threshold, but the association is not accurate enough for diagnosis.
Does stress cause chronic pain?
The review supports interaction between stress physiology and pain sensitivity, especially in experimental pain testing. It cannot establish the causal order by itself.
Is HRV useless for pain?
No. HRV may still be relevant, especially in recovery and intervention studies, but resting HRV alone was not a clean universal marker.
How should stress markers shape chronic-pain research?
Stress physiology is part of chronic primary pain biology. It should inform multidomain treatment and research design, not replace pain assessment or become a simplistic biomarker.
References
- Vyverman J, et al. The stress-pain connection in chronic primary pain: a systematic review and meta-analysis. Neuroscience and Biobehavioral Reviews. 2026;184:106604. doi:10.1016/j.neubiorev.2026.106604
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2-S15. doi:10.1016/j.pain.2010.09.030
- McEwen BS. Protective and damaging effects of stress mediators. New England Journal of Medicine. 1998;338:171-179. doi:10.1056/nejm199801153380307
- Koenig J, Jarczok MN, Ellis RJ, et al. Two-week test-retest stability of the cold pressor task procedure at 2 different temperatures as a measure of pain threshold and tolerance. Pain Practice. 2014;14:E126-E135. doi:10.1111/papr.12142
- Meeus M, Nijs J, Van de Wauwer N, Toeback L, Truijen S. Diffuse noxious inhibitory control is delayed in chronic fatigue syndrome: an experimental study. Pain. 2008;139:439-448. doi:10.1016/j.pain.2008.05.018