
A 2026 PLOS One computational study linked Trichostatin A to 949 predicted targets and 8 machine-learning-selected Alzheimer’s disease core genes, but 100 ns molecular dynamics simulations suggested the compound did not stably bind those candidate proteins.1 That makes the result more useful as an epigenetic network hypothesis than as evidence for a near-term Alzheimer’s drug.
Research Highlights
- 949 TSA targets were predicted: The study integrated ChEMBL, PharmMapper, and SwissTargetPrediction before overlap with Alzheimer’s disease gene datasets.1
- 5 GEO datasets fed the analysis: Batch-corrected Alzheimer’s expression data were used to identify differential genes and modules, including a 295-sample merged training cohort.1
- 130 machine-learning models narrowed the list: The final 8 genes were EFNA1, GABRB2, GABARAPL1, EGR1, CDK5, KCNC2, MET, and GRIA2.1
- Docking did not close the loop: Molecular dynamics suggested TSA may not stably bind the candidate proteins, pushing interpretation toward indirect HDAC1-3/6 regulation.1
- Preclinical support remains separate: Mouse studies have reported TSA-related amyloid, cognition, and behavioral signals, but the 2026 paper itself did not run a treatment experiment.2,3
Histone deacetylases (HDACs) are enzymes that remove acetyl groups from histones and other proteins, changing how tightly DNA is packaged and how genes are expressed. HDAC inhibitors can loosen that regulatory state, which is why they have been studied in cancer, synaptic plasticity, memory, inflammation, and neurodegeneration.
Trichostatin A, often shortened to TSA, is a broad HDAC inhibitor used mostly as an experimental compound. It is not an approved Alzheimer’s therapy.
Broad HDAC inhibition: the biology is powerful but clinically messy. A compound that changes expression across many genes may improve one disease-relevant pathway while disrupting other cell functions.
In Alzheimer’s disease, that risk is especially important because neurons, astrocytes, microglia, endothelial cells, and peripheral immune cells can all respond differently to epigenetic pressure.
949 Predicted Targets Collapsed to 8 Alzheimer’s Core Genes
Ou et al. started with TSA target prediction rather than a biological experiment. ChEMBL contributed 661 targets, PharmMapper contributed 290, and SwissTargetPrediction contributed 102. After deduplication, the study retained 949 nonredundant TSA targets.1
The Alzheimer’s side used 5 Gene Expression Omnibus datasets, differential expression analysis, weighted gene co-expression network analysis, protein-protein interaction filtering, and a 130-model machine-learning screen. The final 8 core genes were:
- EFNA1: an ephrin signaling gene that the paper framed as pathologically upregulated.
- GABRB2 and GRIA2: receptor-related genes tied to inhibitory and excitatory neurotransmission.
- GABARAPL1: an autophagy-related gene relevant to protein handling.
- EGR1: an activity-regulated transcription factor linked to plasticity.
- CDK5: a kinase with relevance to tau phosphorylation and neuronal stress.
- KCNC2: a potassium-channel gene tied to neuronal excitability.
- MET: a receptor tyrosine kinase involved in growth and repair signaling.
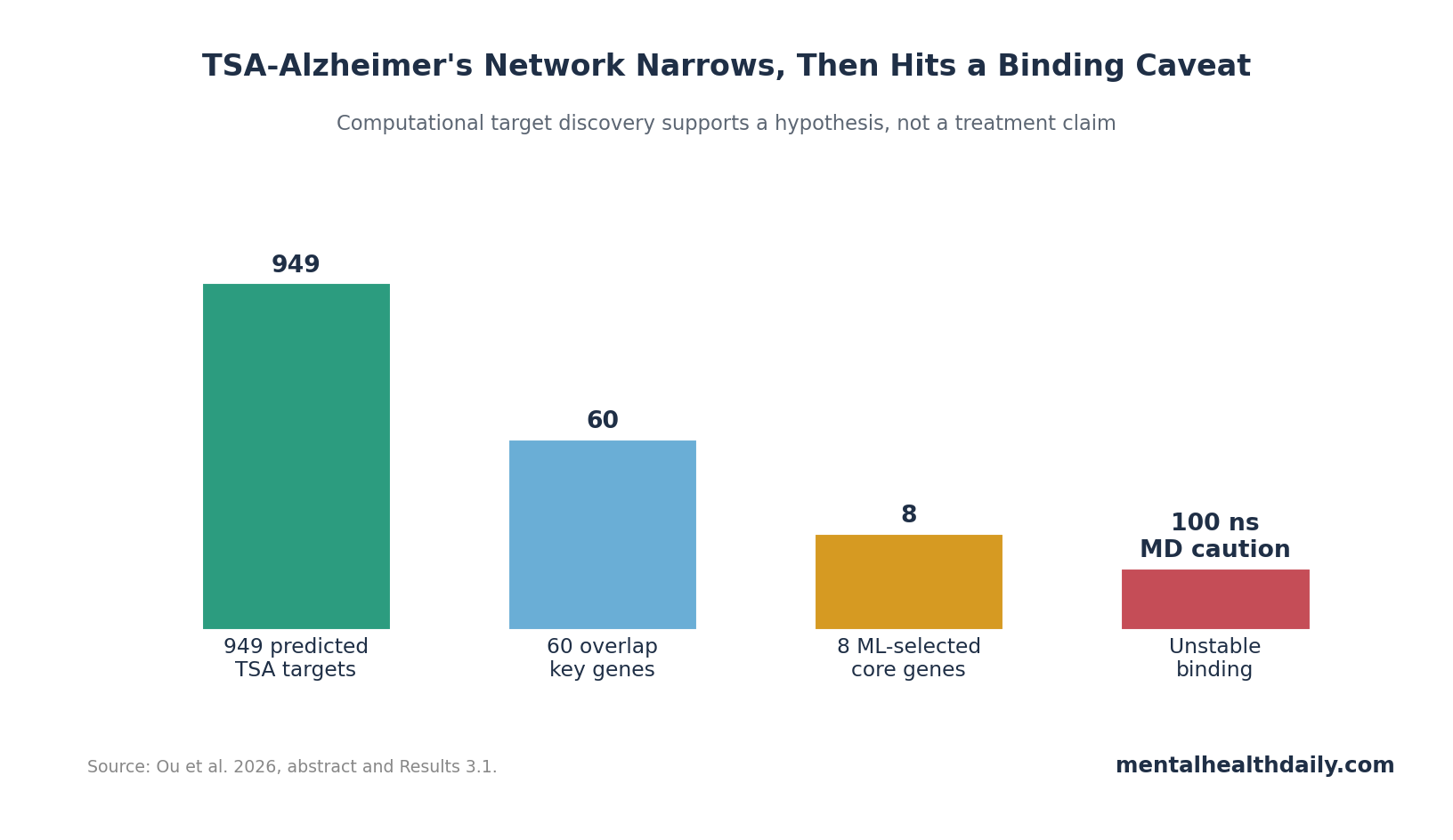
Why the funnel matters: each step reduced a very broad chemical-genomic search space, but each step also added assumptions.
- Target-prediction databases can miss real targets or include false positives.
- Expression datasets can reflect cell-mixture changes rather than disease mechanisms.
- Machine-learning feature selection can overfit when datasets are small or heterogeneous.
The 8-gene list is therefore a priority list for experiments, not a final map of TSA action in the human brain.
The 5 GEO datasets also need clinical humility. Public transcriptomic datasets can combine different brain regions, disease stages, postmortem intervals, diagnostic criteria, and technical platforms. Batch correction helps reduce technical noise, but it cannot make every cohort biologically equivalent. A gene that survives that pipeline is interesting because it recurs across messy data; it is not automatically causal.
The Strongest Result Was Network Direction, Not Target Binding
The paper’s most important calibration is easy to miss. The machine-learning and enrichment results linked TSA to Alzheimer’s-relevant pathways including GABAergic synapse and tau phosphorylation. The candidate genes also fit themes of neurotransmission, synaptic plasticity, tau clearance, and immune-neural crosstalk.1
Then molecular dynamics weakened the direct-binding story. The researchers reported that TSA may not stably bind the candidate proteins across simulation, implying that regulation may occur through upstream HDAC1-3/6 inhibition rather than direct interaction with EFNA1, GABRB2, CDK5, or the other core genes.
Mechanistic read: TSA may act less like a key fitting 8 disease-protein locks and more like an epigenetic lever that changes the expression state of a network. That is plausible for an HDAC inhibitor, but it is also harder to translate into dose, safety, and target-engagement claims.
This distinction keeps the paper from being overread. If a drug directly binds a disease protein, researchers can measure occupancy, affinity, downstream signaling, and dose-response around that protein. If the effect is indirect epigenetic regulation, the relevant evidence has to show which HDAC isoforms are inhibited, which cell types respond, how long the expression shift lasts, and whether the same shift helps or harms synapses, glia, and vascular cells.
Gene-level validation: CDK5 and GRIA2 are plausible Alzheimer’s-relevant genes, but plausibility is not enough.
A follow-up experiment would need 3 checks:
- Expression direction: TSA changes those genes in the expected direction in a relevant cell or animal model.
- Outcome tracking: the gene shift tracks amyloid, tau, synaptic, inflammatory, or cognitive outcomes.
- Pathway dependence: blocking the proposed pathway weakens the effect.
Preclinical Alzheimer’s Data Are Encouraging but Distant
Prior TSA work keeps the idea alive. In APP/PS1 Alzheimer’s-model mice, Su et al. reported that TSA ameliorated Alzheimer’s-related pathology and cognitive deficits by increasing albumin expression and amyloid-β clearance.2 A later APP/PS1 mouse study reported relief of anxiety- and depression-like behaviors after TSA exposure.3
Those papers are not clinical proof. Mouse models compress Alzheimer’s biology into engineered amyloid and behavioral systems. Human Alzheimer’s disease includes age, vascular burden, tau spread, inflammation, sleep, metabolic risk, genetics, and many forms of mixed pathology.
Translation gap: HDAC inhibition can affect many genes at once. That breadth is part of the appeal in a network disease, but it also raises safety and specificity problems.
There is also a dose problem. A compound can look attractive in a mouse or cell model at concentrations that are difficult to deliver safely to the human brain. Before TSA or a related HDAC strategy becomes clinically interesting, researchers would need brain exposure data, tolerability data, and evidence that the desired network shift occurs at doses that do not create unacceptable off-target epigenetic effects.
8 Genes Should Not Be Treated as an Alzheimer’s Blood Test
The 8-gene signature is useful for hypothesis generation. It does not yet function as a validated diagnostic panel. The paper selected genes through computational overlap, model performance, expression patterning, enrichment analysis, and simulation. It did not test a prospective clinical cohort with blinded diagnosis, conversion prediction, or treatment response.
Alzheimer’s biomarkers already include amyloid PET, tau PET, cerebrospinal-fluid amyloid/tau measures, and blood markers such as phosphorylated tau. A gene-expression network would need to prove added value against those established tools before it became clinically relevant.
The comparison standard is demanding because Alzheimer’s disease already has biomarker systems tied to pathology. A transcriptomic signature would need to answer a different question: earlier risk stratification, treatment-response prediction, subtype discovery, or target engagement after an intervention. Without that added use, another gene panel would mostly duplicate existing disease-labeling work.
The cleaner near-term use is experimental triage. If multiple HDAC inhibitors or epigenetic modulators are being compared, the 8-gene network could become one candidate readout among many: does a compound normalize the predicted network, worsen it, or leave it unchanged? That kind of use is narrower than diagnosis but more realistic for a computational paper.
That experimental role also fits the paper’s mixed signal. The target-prediction and machine-learning work point toward a coherent Alzheimer’s network, while the molecular-dynamics step weakens a simple direct-binding mechanism. A good follow-up would treat those 2 findings as a testable tension rather than choosing the more exciting half.
In practical terms, the paper is strongest when read as a shortlist generator for HDAC-focused Alzheimer’s experiments.
Computational Evidence Needs Wet-Lab Confirmation
Evidence-strength note: this was a computational bioinformatics and molecular-simulation study. It can prioritize genes, pathways, and mechanisms for follow-up. It cannot show that TSA improves cognition, slows Alzheimer’s progression, lowers tau burden in humans, or safely changes brain gene expression at tolerable doses.
The next validation steps are straightforward:
- Cell models: test whether TSA changes the 8-gene pattern in neuronal, astrocyte, microglial, and mixed culture systems.
- Animal models: measure whether gene-network changes track amyloid, tau, synaptic markers, inflammation, and behavior.
- Target engagement: confirm which HDAC isoforms and downstream genes actually respond at realistic brain concentrations.
- Safety modeling: assess whether broad epigenetic effects produce unacceptable off-target changes.
Questions About Trichostatin A and Alzheimer’s
Does this paper show TSA treats Alzheimer’s disease?
No. It identifies computational target networks and candidate genes. Treatment efficacy requires biological experiments and clinical trials.
Why does unstable binding matter?
If TSA does not stably bind the candidate proteins, the mechanism is probably indirect. That makes the 8 genes potential downstream markers of HDAC pathway regulation, not confirmed direct drug targets.
Which part is most useful now?
The useful part is the prioritized network: EFNA1, GABRB2, GABARAPL1, EGR1, CDK5, KCNC2, MET, and GRIA2 give experimental labs a focused set of genes to test in Alzheimer’s-relevant models.
References
- Ou C, Chen B, Deng J, Long H. Deciphering the molecular network of Trichostatin A in regulating Alzheimer’s disease screening of core genes and mechanistic investigation based on multidimensional bioinformatics and molecular simulation. PLOS One. 2026;21(4):e0347532. doi:10.1371/journal.pone.0347532
- Su Q, Li T, He PF, et al. Trichostatin A ameliorates Alzheimer’s disease-related pathology and cognitive deficits by increasing albumin expression and Aβ clearance in APP/PS1 mice. Alzheimer’s Research & Therapy. 2021;13:7. doi:10.1186/s13195-020-00746-8
- Su Q, Ren YH, Liu GW, et al. Trichostatin A relieves anxiety- and depression-like symptoms in APP/PS1 mice. Frontiers in Pharmacology. 2024;15:1333235. doi:10.3389/fphar.2024.1333235
- Kumar V, Kundu S, Singh A, Singh S. Understanding the role of histone deacetylase and their inhibitors in neurodegenerative disorders. Current Neuropharmacology. 2022;20(1):158-178. doi:10.2174/1570159×19666210609160017
- Ganai SA, Ramadoss M, Mahadevan V. Histone deacetylase inhibitors: emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Current Neuropharmacology. 2016;14(1):55-71. doi:10.2174/1570159×13666151021111609