
The endogenous opioid system has been a candidate target for novel anxiety treatments, but a 2026 placebo-controlled crossover fMRI study in 38 healthy volunteers ran opposite the hypothesis: instead of increasing threat-related distress and amygdala reactivity, 50 mg oral naltrexone reduced subjective distress during cognitive reappraisal (p = 0.044, d = 0.57) and shifted ventromedial prefrontal cortex (vmPFC) responses to fearful faces.1
Research Highlights
- Hypothesis was directional and got reversed: the prediction was that 50 mg oral naltrexone would increase distress ratings during emotional reappraisal vs. placebo. Distress was actually lower under naltrexone (mean placebo −11.24 vs. naltrexone −9.08; t(37) = 2.08, p = 0.044, d = 0.57).1
- Reappraisal effectiveness was preserved. The instruction main effect (regulate > attend negative) was large and unchanged across drug conditions: F(1,37) = 17.63, p < 0.001, d = 1.31. Naltrexone didn’t impair participants’ ability to reappraise — it shifted the affective baseline.1
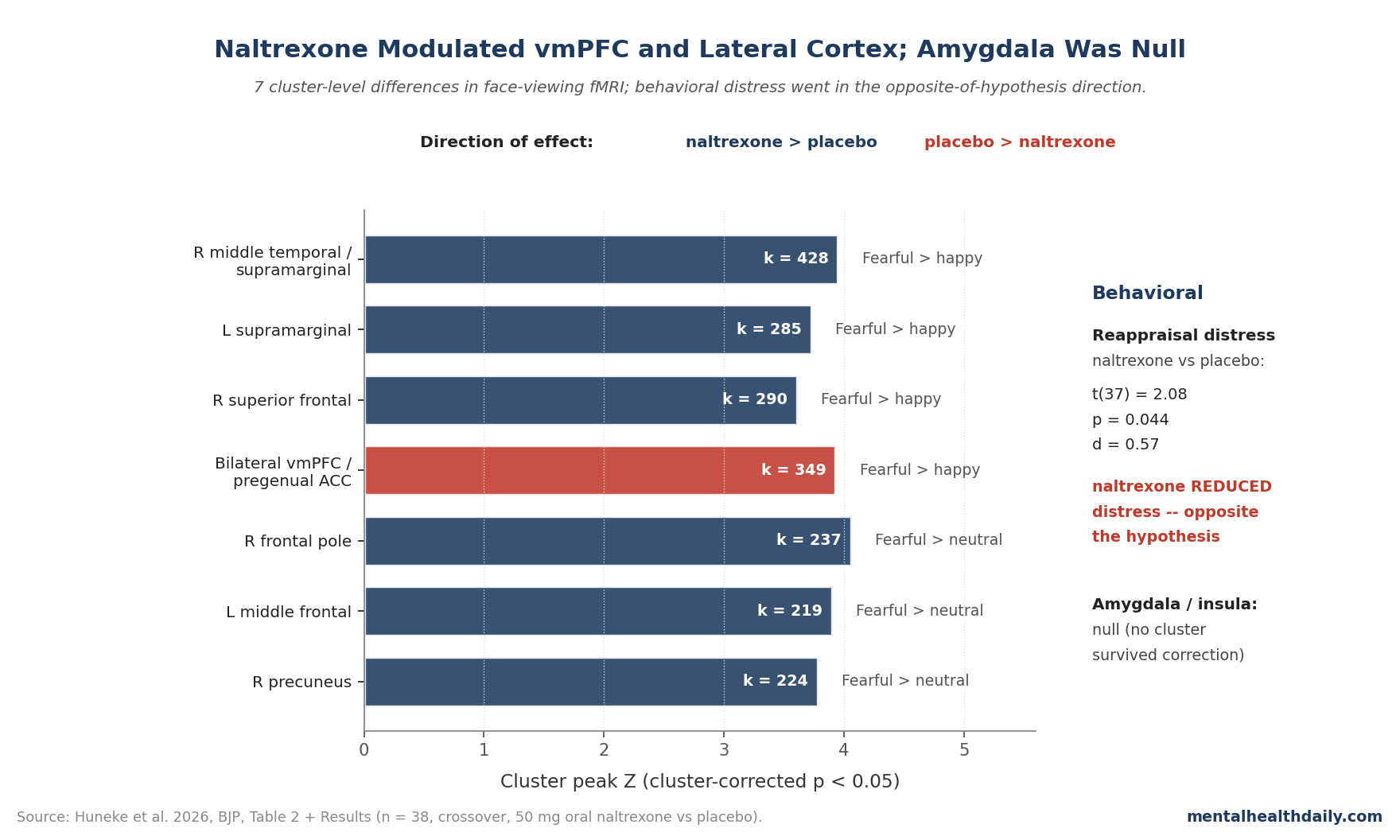
- vmPFC and ACC tracked the drug. In the fearful > happy contrast, the placebo > naltrexone comparison showed bilateral vmPFC/pregenual anterior cingulate activation (k = 349, Z = 3.93, p = 0.005). Naltrexone > placebo for fearful > happy showed right middle temporal/supramarginal (k = 428, Z = 3.95, p = 0.001) and bilateral supramarginal/superior frontal clusters.1
- Amygdala and insula were not differentially modulated. Both regions activated robustly to faces in both conditions, but no between-condition cluster survived correction in those ROIs — the more limbic-anchored prediction was a null, even though the cortical regulation circuit wasn’t.1
- Reaction times were slower under naltrexone than placebo at trend level (F(1,37) = 3.62, p = 0.065). Accuracy showed a valence main effect (fearful > neutral, p = 0.040) but no drug interaction.1
Opioid-system rationale: anxiety disorders affect roughly 30% of adults across the lifespan, and the SSRI/SNRI/CBT pipeline leaves a substantial residual symptom burden — motivating the search for mechanism-novel pharmacology.2
Mu-opioid antagonists became a candidate target on theoretical grounds: animal work shows central-amygdala mu-opioid receptors gate anxiolytic responses,3 and human pharmacology suggests endogenous opioids modulate social-pain processing, hedonic responses, and stress-related affect.4
The Huneke team’s a priori hypothesis sat on top of that frame: blocking mu-opioid receptors should remove an endogenous stress buffer, increasing subjective distress and amygdala reactivity to threat. The study was designed to validate that mechanism in healthy volunteers as a prerequisite to anxiety-disorder trials.
What it found pushes the opposite direction on subjective ratings and a cortical-regulation pattern that wasn’t predicted.
40 Recruited, 38 Analyzed, 50 mg Oral Naltrexone vs. Placebo
The trial enrolled healthy volunteers without psychiatric diagnoses (n = 40 randomized; 38 included in the fMRI analysis after one withdrawal and one exclusion). Each participant received 50 mg oral naltrexone or matched placebo in a counterbalanced double-blind crossover design, with sessions spaced for adequate washout.1
Drug administration preceded fMRI scanning by 1 hour to allow plasma peak coverage of the scanning window.
Two tasks were run in each scan session. The cognitive emotional reappraisal task tested explicit threat regulation: participants viewed aversive images and either passively attended or actively reappraised them in less threatening terms.
This task is well-validated for measuring the dlPFC/vlPFC engagement that downregulates amygdala threat responses.5 The face-viewing task tested implicit threat processing: participants viewed fearful, angry, happy, and neutral faces under low-task-demand conditions while incidental neural responses were tracked.
Subjective distress was rated in real time during reappraisal blocks. Whole-brain fMRI analyses used cluster-based correction (Z > 2.3, cluster-corrected p < 0.05).
Naltrexone Reduced Reappraisal Distress — Opposite the Prediction
The behavioral data alone are striking enough to merit attention. The reappraisal task generated the expected large instruction effect: distress was substantially lower in regulate blocks than attend blocks (F(1,37) = 17.63, p < 0.001), with mean differences of about −3.4 ratings on the 0–100 scale (t(37) = 4.20, p < 0.001, d = 1.31).1
Reappraisal worked, exactly as the literature would predict.
The drug condition main effect went the unexpected direction. Across both attend and regulate blocks, distress was lower under naltrexone than placebo (mean placebo −11.24, 95% CI −15.70 to −6.76; mean naltrexone −9.08, 95% CI −13.20 to −4.98; t(37) = 2.08, p = 0.044, d = 0.57). The hypothesis predicted the opposite.
This isn’t a huge effect — d = 0.57 is solidly in the small-to-medium range, and a 38-participant sample isn’t powered to chase smaller effects with confidence. But the direction matters.
A theory predicting opioid antagonism removes a stress buffer should not produce reduced distress under blockade. Either the buffer hypothesis is wrong as stated, or naltrexone in healthy volunteers under acute single-dose conditions does something the buffer model doesn’t capture — possibly altering how negative stimuli are appraised in the first place rather than how they’re regulated downstream.
vmPFC Activated Less for Fearful Faces Under Naltrexone
The face-viewing task generated the most interpretable cluster-level findings, and they’re directional opposites in two contiguous regions of the medial prefrontal cortex.1
In the fearful > happy contrast, the placebo condition produced significantly more activation than naltrexone in bilateral vmPFC extending to pregenual anterior cingulate cortex (k = 349 voxels, Z = 3.93, p = 0.005). Parameter-estimate inspection traced this to reduced deactivation for happy faces under naltrexone — meaning placebo participants suppressed vmPFC for happy faces more than naltrexone participants did, which inverted the fearful-vs-happy comparison.
In the same fearful > happy contrast, naltrexone produced significantly more activation than placebo in three lateral cortical clusters: right middle temporal/posterior supramarginal gyrus (k = 428, Z = 3.95, p = 0.001), right superior frontal gyrus (k = 290, Z = 3.61, p = 0.016), and left supramarginal gyrus (k = 285, Z = 3.73, p = 0.017).
For fearful > neutral, naltrexone > placebo produced clusters in right frontal pole, right precuneus, and left middle frontal gyrus. The precuneus effect was driven by a smaller deactivation for fearful faces under naltrexone.
Amygdala and Insula Were Null; vmPFC Modulation Was Not
One read of the paper would say “subcortical-circuit null, cortical-regulation finding.” That separation matters because the original mechanism hypothesis was specifically subcortical: mu-opioid receptors in the amygdala gating reactivity to threat.3
Bilateral amygdala and bilateral insula activated robustly to faces in both drug conditions. Neither showed differentiation by drug at the corrected whole-brain or ROI threshold.1
For the specific subcortical buffering hypothesis — “remove the opioid brake on the amygdala and reactivity to threat goes up” — the amygdala data are a real null.
Where things become interesting is upstream. vmPFC and pregenual ACC are core nodes of the appraisal-and-evaluation circuit, and they’re where naltrexone produced the largest single cluster (k = 349 voxels).
The directional pattern — less vmPFC for fearful relative to happy under placebo, less of that suppression under naltrexone — suggests opioid antagonism shifts the relative weighting of valence in medial prefrontal evaluation rather than altering subcortical reactivity per se. That’s a different mechanism story than the one the trial was built to test.
Why This Reframes the Anxiety-Drug Hypothesis
If the result were truly null across the board, it would close the door cleanly on opioid antagonism as an anxiety mechanism. It isn’t, and it doesn’t.
The actual pattern updates the mechanism story in three independent ways — the stress-buffer framing, the direction of the behavioral effect, and the case for selective subtype targeting.
The “stress buffer” framing is wrong as stated. Acute mu-opioid blockade in healthy volunteers reduced rather than increased subjective distress and didn’t elevate amygdala reactivity. That’s incompatible with the most common mechanistic argument for opioid antagonists in anxiety treatment, which assumes endogenous opioids dampen threat reactivity and blocking them removes the dampening.
The anti-anxiety direction of effect could be real. A reduction in distress under naltrexone, even if small (d = 0.57), is not what stress-buffer theory predicts. It’s at least consistent with mu-opioid antagonism altering reward–threat valuation in a way that changes appraisal, more than downstream reactivity.
This is closer to the mechanism by which low-dose naltrexone has been investigated in chronic pain, fibromyalgia, and inflammation-related affective conditions, where the effect signal also runs through valence processing.6
Selective subtype targeting may still be on the table. Naltrexone is a non-selective antagonist (mu, kappa, delta). Kappa antagonists in particular have been pursued for anxiety and depression on a different mechanism story — kappa-opioid signaling drives stress-related dysphoria, and kappa antagonism may produce stress-resilience effects that mu-targeted compounds don’t.7 The Huneke design can’t separate these.
How This Lines Up with Prior Opioid-Antagonism fMRI Work
This isn’t the first pharmacological-fMRI study of opioid antagonism in healthy volunteers, and the prior literature has been mixed.8,4
Eikemo et al. found that opioid antagonism modulated the hedonic response to pleasant tastes and music but produced inconsistent effects on threat-related circuits. Chelnokova et al. showed selective mu-receptor effects on social reward processing.
Saanijoki et al. demonstrated mu-opioid release during exercise-induced affect change. None of these prior studies replicated the simple “block opioids, threat reactivity goes up” pattern that mu-stress-buffer theory predicted.
The Huneke result fits that broader pattern. Endogenous opioid signaling appears more relevant to valence and reward processing than to subcortical threat reactivity per se.
The clinical implication is that anxiety drugs targeting endogenous opioid systems should be evaluated through reward-and-valence outcome measures, not solely through threat-circuit engagement assays.
Where the Huneke Design Limits Inference
The trial is high-quality on the design axes that matter most — double-blind, placebo-controlled, within-subject crossover, prespecified contrasts, cluster-corrected fMRI — but four design features bound what can be concluded.
Acute single dose. Most clinical opioid-antagonist research uses chronic dosing where receptor adaptation may matter. The Huneke 1-hour pretreatment captures peak plasma but not the steady-state pharmacology relevant to clinical anxiety treatment.
Healthy volunteers. Anxiety patients have elevated threat-circuit reactivity at baseline. The pharmacology may interact with disordered substrate differently than with normal-range substrate.
The trial doesn’t address that question and doesn’t claim to.
Sample size. Within-subject n = 38 is well-powered for medium effects (d ≈ 0.5) but underpowered for small effects. The amygdala null is most defensible if amygdala drug effects are medium or larger; smaller effects could be present but undetectable.
Non-selective antagonism. Naltrexone blocks mu, kappa, and delta receptors. Subtype-selective compounds — kappa antagonists, mu-selective tools — may produce different patterns and aren’t ruled out by this result.
Questions About Naltrexone and Threat Processing
What did the study actually find?
Naltrexone reduced subjective distress during emotional reappraisal (the opposite of the prediction) at p = 0.044, d = 0.57. Reappraisal effectiveness was preserved (instruction effect F(1,37) = 17.63, p < 0.001, d = 1.31).
At the brain level, vmPFC/pregenual ACC activated less for fearful > happy under naltrexone; lateral cortical regions (right middle temporal/supramarginal, right superior frontal, precuneus) activated more. Amygdala and insula were not differentially modulated.
Was this a null result?
Partly. The amygdala-and-insula prediction (the core of the mu-opioid stress-buffer hypothesis) was a null.
The behavioral and cortical-regulation findings were not. Calling the whole study a clean null misses that the directional behavioral effect was opposite the hypothesis — naltrexone produced lower distress, not higher.
Does this support naltrexone for anxiety?
It doesn’t recommend it, and it doesn’t rule it out. The reduction in distress is in the anti-anxiety direction, but it’s a single small healthy-volunteer signal under acute dosing — far below the threshold for clinical recommendation.
What it does is shift the mechanism story away from subcortical threat-buffering and toward valence/appraisal modulation, which has implications for how future opioid-system anxiety drugs should be evaluated.
Why might naltrexone reduce distress in healthy volunteers?
The leading interpretation is that mu-opioid antagonism alters reward–valence processing rather than subcortical threat reactivity. That’s consistent with prior pharmacological-fMRI work showing opioid effects on hedonic processing of food, music, and social reward but inconsistent threat-circuit effects.
The vmPFC pattern in this study supports an appraisal-shift mechanism rather than a threat-suppression one.
Was the amygdala result really negative?
Yes, at the corrected whole-brain and ROI levels for both tasks. Amygdala activated robustly to faces in both drug conditions but didn’t differ between conditions.
The amygdala-specific buffering hypothesis is not supported in this dataset.
What evidence would change the mechanism call?
Chronic-dosing pharmacological-fMRI in clinical anxiety populations would test whether the acute healthy-volunteer pattern generalizes — the receptor-level adaptations that come with weeks of dosing aren’t captured in a 1-hour pretreatment. Subtype-selective compounds, particularly kappa-opioid antagonists with a different mechanism story for stress and dysphoria, should run through the same paradigms before opioid-system targets are written off as a class.
And reward/valence-focused outcome measures should be added to opioid-system trials alongside the threat-circuit engagement assays, given that prior pharmacology (Eikemo, Chelnokova, Saanijoki) consistently points to valence rather than threat as the more sensitive readout.
How does this compare to standard anxiety treatments?
SSRIs, SNRIs, and CBT have effect sizes comparable to or larger than what this single-dose pharmacology produced, with much larger evidence bases.2 The Huneke result doesn’t change clinical practice.
It informs upstream mechanism research and drug-development decision-making.
References
- Huneke NTM, van Steenbergen H, Fagan HA, et al. Effects of opioid antagonism on functional MRI correlates of explicit and implicit threat processing in healthy volunteers. British Journal of Psychiatry. 2026. doi:10.1192/bjp.2026.10605
- Bandelow B, Reitt M, Röver C, Michaelis S, Görlich Y, Wedekind D. Efficacy of treatments for anxiety disorders: a meta-analysis. International Clinical Psychopharmacology. 2015;30(4):183–192. doi:10.1097/YIC.0000000000000078
- Burghardt PR, Wilson MA. Microinjection of naltrexone into the central, but not the basolateral, amygdala blocks the anxiolytic effects of diazepam in the plus maze. Neuropsychopharmacology. 2006;31(6):1227–1240. doi:10.1038/sj.npp.1300864
- Eikemo M, Loseth GE, Johnstone T, Gjerstad J, Willoch F, Leknes S. Sweet taste pleasantness is modulated by morphine and naltrexone. Psychopharmacology. 2016;233(21–22):3711–3723. doi:10.1007/s00213-016-4403-x
- Buhle JT, Silvers JA, Wager TD, et al. Cognitive reappraisal of emotion: a meta-analysis of human neuroimaging studies. Cerebral Cortex. 2014;24(11):2981–2990. doi:10.1093/cercor/bht154
- Younger J, Parkitny L, McLain D. The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clinical Rheumatology. 2014;33(4):451–459. doi:10.1007/s10067-014-2517-2
- Carlezon WA Jr, Krystal AD. Kappa-opioid antagonists for psychiatric disorders: from bench to clinical trials. Depression and Anxiety. 2016;33(10):895–906. doi:10.1002/da.22500
- Chelnokova O, Laeng B, Eikemo M, et al. Rewards of beauty: the opioid system mediates social motivation in humans. Molecular Psychiatry. 2014;19(7):746–747. doi:10.1038/mp.2014.1