
A 2026 review ties the Parkinson’s disease genes PINK1 and parkin to damaged-mitochondria cleanup, from damage sensing and ubiquitin tagging to autophagy recruitment and lysosomal degradation.1 That shifts “mitochondrial dysfunction” into specific failure points: tagging, extraction, transport, autophagosome recruitment, and degradation.
Research Highlights
- 18 PARK loci frame inherited risk: PARK2 encodes parkin and PARK6 encodes PINK1, 2 recessive parkinsonism genes tied to mitochondrial cleanup.
- Cases nearly tripled in recent data: the review cites a meta-analysis reporting almost a triplication of Parkinson’s disease cases between 2010 and 2023.
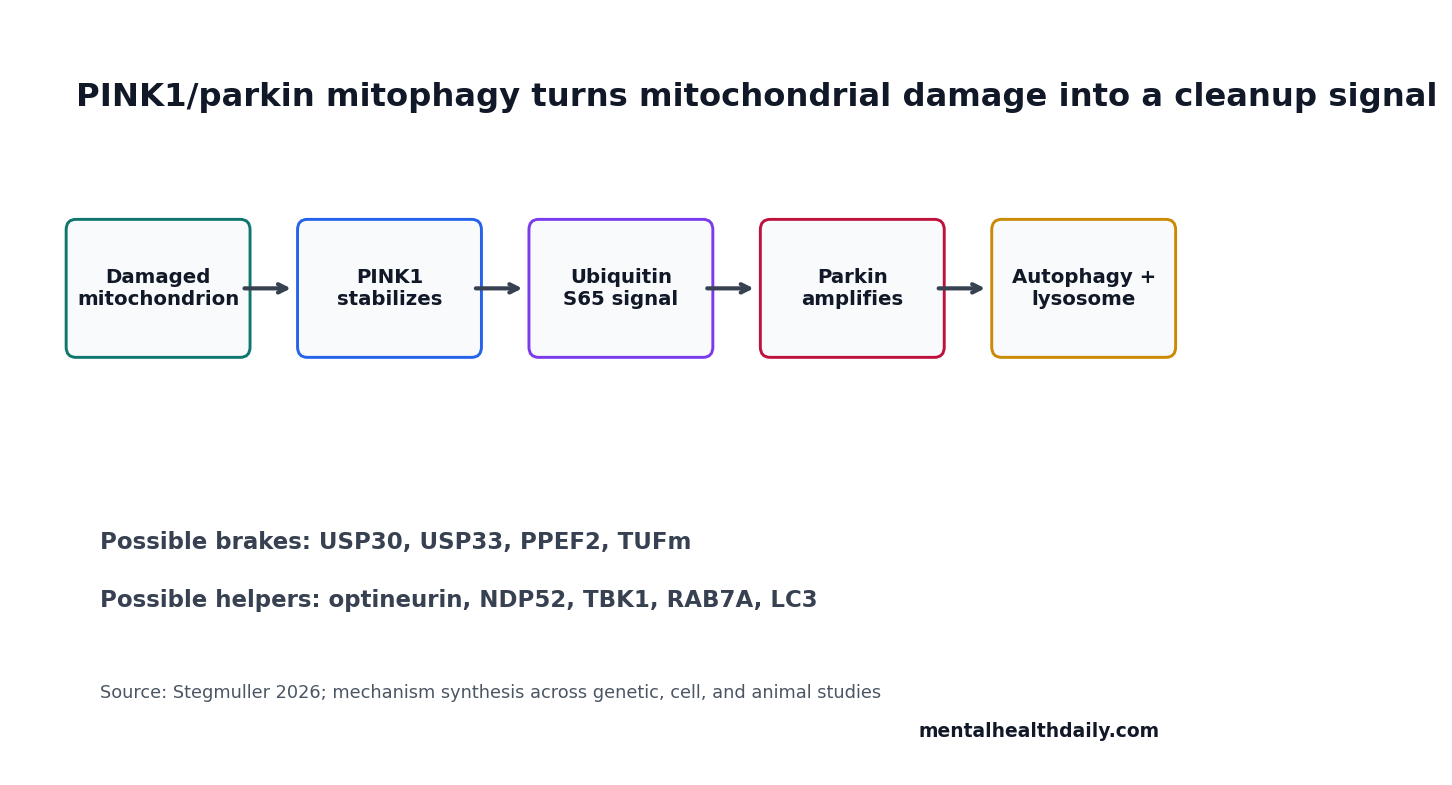
- PINK1 starts the damage signal: damaged mitochondria stabilize PINK1, which phosphorylates ubiquitin at S65 and helps recruit parkin.
- Parkin expands the ubiquitin mark: parkin modifies outer-mitochondrial and stress-related substrates including PHB2, BAK, MIRO, and MITOL-linked proteins.
- Therapeutic ideas remain early: USP30 inhibitors, ROCK2 inhibitors, PINK1 activity tuning, and mitophagy probes are research tools, not 2026 clinical treatments.
Mitophagy is selective autophagy of mitochondria: the cell identifies damaged mitochondria, encloses them in an autophagosome, and sends them for lysosomal degradation. In neurons, this is especially important because axons are long, energy demand is high, and damaged mitochondria can create oxidative stress.
PINK1 is a mitochondrial kinase, an enzyme that adds phosphate groups to target proteins. Parkin is an E3 ubiquitin ligase, an enzyme that attaches ubiquitin tags to proteins so the cell can route them toward repair, relocation, or degradation.
PINK1 Accumulates When Mitochondria Lose Membrane Potential
Healthy mitochondria import and rapidly degrade PINK1. When a mitochondrion becomes damaged or depolarized, PINK1 accumulates on the outer mitochondrial membrane and becomes active through autophosphorylation.
PINK1 then phosphorylates ubiquitin at S65 and helps recruit parkin. Parkin ubiquitinates proteins on the mitochondrial surface, expanding the signal that this mitochondrion needs disposal. That sequence turns local mitochondrial damage into a visible cellular cleanup tag.
Reader translation: PINK1 acts like a damage sensor. Parkin acts like a tag amplifier. The autophagy system then reads the tag and starts clearing the damaged organelle.
Parkin Substrates Show That Mitophagy Is Not One Simple Switch
Stegmuller summarizes newer work showing that parkin modifies more than a generic mitochondrial surface. Prohibitin 2 can become a parkin target and increase affinity for LC3, a protein associated with autophagosome membranes. BAK can be ubiquitinated in a way that limits apoptosis while allowing mitochondrial clearance.
MIRO proteins help move mitochondria along microtubules in long axons. Their interaction with PINK1 and parkin matters because neurons cannot simply wait for damaged mitochondria to drift back to the cell body. Local transport, local translation, and local tagging all shape whether cleanup happens in time.
- PHB2: can connect inner-mitochondrial damage exposure to LC3 recognition.
- BAK: sits near the apoptosis-vs.-mitophagy decision.
- MIRO1/2: link mitochondrial movement to parkin recruitment and clearance.
- MITOL: can regulate parkin degradation and relocate away from damaged mitochondria.
Deubiquitinases Can Brake the Cleanup Signal
Deubiquitinases (DUBs) remove ubiquitin tags. If parkin adds cleanup marks, DUBs can trim or erase them. That makes DUBs attractive drug targets because inhibiting the right DUB might strengthen mitophagy without forcing every upstream step.
USP30 is the clearest example in the review. It sits on mitochondria and opposes PINK1/parkin-related ubiquitin signaling. Peptide and small-molecule USP30 inhibition increased phosphorylated ubiquitin or mitophagy-linked events in experimental systems. USP14 and USP33 also appear in the regulatory landscape, though their effects depend on model and pathway context.
Therapeutic boundary: blocking a DUB in cells is not the same as treating Parkinson’s disease. It identifies a lever. Human therapy would need brain exposure, safety, target engagement, durability, and evidence that more mitophagy helps the right neurons at the right disease stage.
PINK1 Regulation Extends Beyond Parkin Recruitment
Newer PINK1 work adds several layers. The review describes neuron-specific transport of pink1 mRNA along mitochondria, allowing local PINK1 production in long neuronal processes. SMAD3 can be phosphorylated by PINK1 and regulate pink1 expression, creating a feedback route after mitochondrial damage.
TOM7, OMA1, AMBRA1, ATAD3A, PPEF2, and TUFm also enter the pathway. These names can look like alphabet soup, but the categories are understandable:
- Import and cleavage controls: TOM7, OMA1, PARL, and related proteases influence whether PINK1 is imported, stabilized, or degraded.
- Autophagy-initiation controls: AMBRA1, optineurin, NDP52, TBK1, and ULK-related complexes help recruit or organize autophagy machinery.
- Phosphate controls: PPEF2 can remove phosphate from phosphorylated ubiquitin, countering the PINK1 signal.
- Translation and stress controls: TUFm and pink1 mRNA localization connect mitochondrial translation and local neuronal response.
Other PARK Genes Converge on Mitophagy
PINK1 and parkin are central, but they are not isolated. The review connects alpha-synuclein, LRRK2, DJ-1, VPS35, and FBXO7 to mitochondrial quality control or autophagy-linked processes. That convergence helps explain why Parkinson’s genetics can look diverse while still pointing toward overlapping cellular failure modes.
Genetic calibration: PARK2 and PARK6 mutations are most directly tied to recessive early-onset parkinsonism. Common late-onset Parkinson’s disease is usually not a simple PINK1/parkin disease. The pathway is still relevant because sporadic disease can stress similar mitochondrial, lysosomal, ubiquitin, and autophagy systems.
LRRK2 mutations can impair mitophagy-related recruitment of optineurin and RAB proteins. DJ-1 appears to act downstream of PINK1/parkin activation in some models. VPS35 variants affect mitochondrial membrane potential and recruitment. These links turn mitophagy into a shared pathway rather than a narrow recessive-gene footnote.
Mitophagy Drug Ideas Are Plausible but Early
Several strategies appear attractive: inhibit USP30, tune PINK1 kinase activity, increase parkin recruitment, alter ROCK2 signaling, improve lysosomal handling, or use better probes to identify cells with failed mitophagy. Each strategy has a different risk profile.
Evidence-strength note: Stegmuller’s paper is a narrative mechanistic review, not a clinical trial or systematic meta-analysis. Its value is synthesis across cell biology, genetics, animal models, and emerging drug-target work. It cannot establish that a mitophagy drug will slow Parkinson’s disease in people.
The 2026 takeaway is still actionable for interpretation. “Mitochondrial dysfunction” is too vague. PINK1/parkin biology asks a more specific question: which step of damaged-mitochondria recognition, ubiquitin marking, autophagosome recruitment, or lysosomal clearance is failing, in which neuron type, at which stage?
The Therapeutic Question Is Which Cleanup Step to Tune
Mitophagy is not one drug target. It is a sequence of checkpoints. A therapy aimed at PINK1 kinase activity would sit near the damage-sensing step. A USP30 inhibitor would sit near the ubiquitin-signal braking step. A lysosomal enhancer would sit later, after damaged mitochondria have already been tagged and enclosed.
Early checkpoint: PINK1 stabilization and S65 ubiquitin phosphorylation ask whether the neuron recognizes mitochondrial damage quickly enough. Failure here means the cleanup program may never start.
Middle checkpoint: parkin recruitment, mitochondrial substrate ubiquitination, and autophagy-receptor binding ask whether the damage mark becomes large and organized enough for autophagosome machinery to read it.
Late checkpoint: lysosomal delivery and degradation ask whether a tagged mitochondrion actually gets broken down. Strengthening early tagging would not solve a downstream lysosomal bottleneck, and pushing late degradation would not fix a missing PINK1/parkin signal.
This is why the review’s long list of proteins separates possible failure modes. A common late-onset Parkinson’s patient, a PARK2 carrier, and a cell model with USP30 overactivity may all be described as having mitophagy stress, but the intervention logic is different in each case.
The disease-stage problem is just as important. Early neurons with recoverable mitochondrial quality-control stress might benefit from more efficient tagging or lysosomal handling. Late neurons with severe axonal degeneration, protein aggregation, and inflammatory stress may not respond to the same lever. A useful mitophagy therapy would therefore need a biomarker of pathway engagement plus a plausible target in a dish.
That is where PINK1/parkin biology becomes clinically disciplined. It pushes drug development toward specific readouts: phosphorylated ubiquitin, parkin recruitment, mitochondrial substrate turnover, autophagosome formation, lysosomal clearance, and neuron survival. Without those step-specific readouts, “improves mitochondrial function” is too vague to separate a real Parkinson’s therapy from a generic cell-stress result.
Genetic context changes interpretation: a recessive PARK2 or PARK6 case starts closer to the pathway than a typical sporadic case, so the same biomarker panel may not mean the same thing across patients.
Clinical translation would need to stratify by genetics, age at onset, disease stage, and dopaminergic-neuron vulnerability before treating mitophagy as one uniform target.
Cell biology also sets the safety problem: more mitophagy is not automatically better. Neurons need to remove damaged mitochondria, but they also need enough working mitochondria to maintain axons, synapses, calcium handling, and neurotransmission. A therapy that pushes clearance without supporting replacement could trade one stress for another.
Measurable pathway parts: PINK1 accumulation, ubiquitin phosphorylation, parkin substrate marking, receptor recruitment, and lysosomal degradation can each become a separate experiment.
Parkinson’s drug development needs that granularity because a clinical failure at one step should not invalidate the whole mitophagy hypothesis.
Questions About PINK1, Parkin, and Mitophagy
Are PINK1 and parkin the cause of most Parkinson’s disease?
No. PARK2 and PARK6 are important recessive parkinsonism genes, but most Parkinson’s disease is not caused by simple mutations in those 2 genes.
What does failed mitochondrial cleanup mean?
It means damaged mitochondria are not recognized, tagged, transported, enclosed, or degraded efficiently enough, leaving neurons exposed to energy failure and stress signals.
Are USP30 inhibitors Parkinson’s treatments now?
No. USP30 inhibition is a mechanistically interesting way to strengthen mitophagy signals in experimental systems. Clinical usefulness remains unproven.
References
- Stegmuller J. Novel insight into PINK1/parkin-associated autophagy implicated in Parkinson disease. Translational Neuroscience. 2026;17(1):20250386. https://doi.org/10.1515/tnsci-2025-0386
- PubMed search: PINK1 parkin mitophagy Parkinson disease review. https://pubmed.ncbi.nlm.nih.gov/?term=PINK1+parkin+mitophagy+Parkinson+disease+review
- PubMed search: USP30 inhibitor mitophagy Parkinson disease PINK1 parkin. https://pubmed.ncbi.nlm.nih.gov/?term=USP30+inhibitor+mitophagy+Parkinson+disease+PINK1+parkin
- PubMed search: LRRK2 DJ-1 VPS35 mitophagy Parkinson disease. https://pubmed.ncbi.nlm.nih.gov/?term=LRRK2+DJ-1+VPS35+mitophagy+Parkinson+disease