
A 2026 structured review of autoimmune drug-resistant focal epilepsy argues for a narrow but clinically important claim: immune mechanisms can make seizures refractory in selected patients, but the evidence supports targeted testing and immunotherapy triage rather than treating every medication-resistant seizure disorder as autoimmune.
Research Highlights
- Autoimmune epilepsy is a subset: Kunecki et al. estimated that autoimmune causes may account for 10-20% of acute-onset epilepsy cases, while neuronal autoantibodies appear in fewer than 10% of focal drug-resistant epilepsy cases.
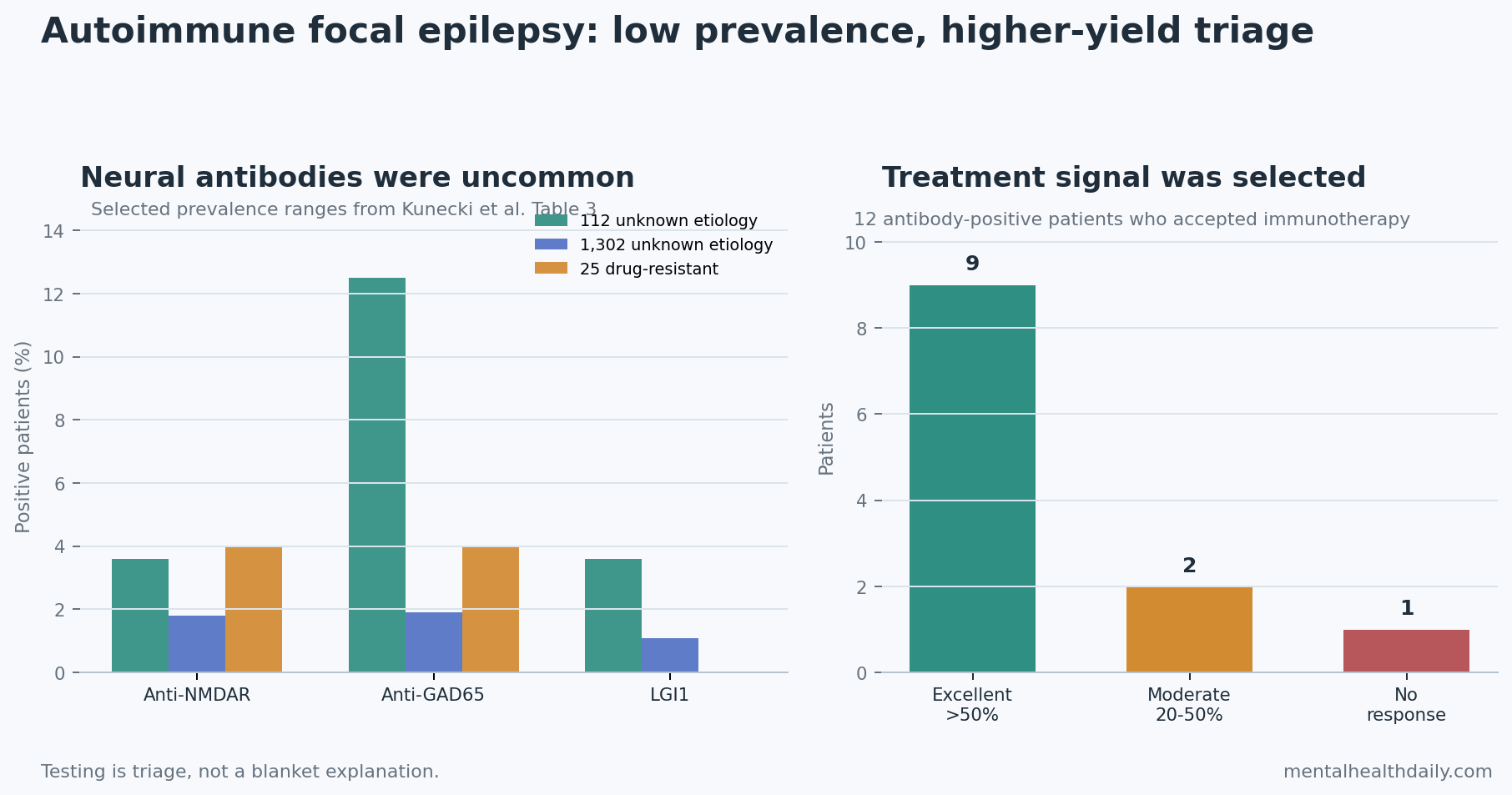
- Antibody rates are low but not zero: summarized studies reported anti-NMDAR positivity around 1.8-4%, anti-GAD65 around 1.9-12.5%, and LGI1 around 0-3.6%, depending on the population.
- Immunotherapy response is promising in selected patients: in the key antibody-positive treatment cohort, 9 of 12 treated patients had more than 50% seizure reduction, 2 had 20-50% reduction, and 1 did not respond.
- Mechanisms converge on brain inflammation: blood-brain barrier disruption, astrocyte activation, IL-1β, IL-6, TNF-α, mTOR signaling, and efflux transporters offer plausible routes from immune activation to seizure persistence.
- Evidence strength remains limited: the review covered 4 immunotherapy categories, case reports, registry data, clinical trial records, and biomarker studies, but no formal risk-of-bias assessment or meta-analysis of immunotherapy response.
Drug-resistant epilepsy means seizures continue after 2 appropriately chosen, tolerated, and adequately dosed antiseizure medications. That definition is useful because it marks the point where clinicians should stop repeating ordinary medication substitutions and start asking whether surgery, neuromodulation, genetics, structural lesions, inflammation, or immune-mediated disease is being missed.
Autoimmune epilepsy is epilepsy driven or amplified by immune attack on the brain, often through neuronal surface antibodies, inflammatory cytokines, blood-brain barrier injury, or autoimmune encephalitis. The hard part is calibration: autoimmune epilepsy is real and sometimes treatable, but it is not the default explanation for all refractory focal seizures.
Autoimmune Clues Should Trigger Triage, Not Reflex Immunotherapy
Kunecki et al. reviewed immune mechanisms in drug-resistant focal epilepsy and framed the autoimmune signal around several pathways. The paper highlighted blood-brain barrier dysfunction, which means the vascular boundary that normally limits immune and blood-protein entry into brain tissue becomes leakier. Once albumin, antibodies, complement, and inflammatory mediators enter brain tissue, astrocytes and endothelial cells can shift toward a proinflammatory state.
That inflammatory loop can plausibly support seizures in 2 ways:
- More excitable brain tissue: cytokines such as IL-1β, IL-6, and TNF-α can alter synaptic signaling, glial activation, and local network stability.
- Less drug penetration: blood-brain barrier injury can upregulate efflux transporters such as P-glycoprotein, which can pump some antiseizure medications away from brain tissue and contribute to pharmacoresistance.
mTOR signaling adds a second route. The mammalian target of rapamycin pathway regulates cell growth, protein synthesis, synaptic plasticity, autophagy, and neuronal development. Hyperactivation has been linked to abnormal axonal growth, altered neurogenesis, and epileptogenesis, especially in developmental epilepsies and tuberous sclerosis spectrum biology.
The practical interpretation is selective workup. A patient with focal seizures, rapid onset, cognitive or psychiatric changes, inflammatory CSF, MRI limbic signal, unexplained status epilepticus, personal autoimmune disease, or neuronal antibodies belongs in a different diagnostic lane than a patient with long-standing focal epilepsy from a stable structural lesion and no immune red flags.
Neuronal Autoantibodies Are Useful Because They Are Specific, Not Common
Neuronal autoantibodies are antibodies directed at brain proteins involved in synaptic signaling, ion-channel function, or intracellular neuronal targets. The review emphasized N-methyl-D-aspartate receptor antibodies (anti-NMDAR), glutamic acid decarboxylase 65 antibodies (anti-GAD65), leucine-rich glioma-inactivated 1 antibodies (LGI1), contactin-associated protein-like 2 antibodies (CASPR2), and GluR3 antibodies against an AMPA receptor subunit.
Prevalence numbers keep the claim honest. In the review’s antibody table, a 112-person epilepsy-of-unknown-etiology study reported anti-NMDAR antibodies in 3.6%, anti-GAD65 in 12.5%, LGI1 in 3.6%, and thyroid peroxidase antibodies in 13.4%. A larger 1,302-person synthesis reported lower neural-antibody rates: anti-NMDAR 1.8%, anti-GAD65 1.9%, LGI1 1.1%, and onconeural antibodies 0.2%. A 25-person Egyptian drug-resistant epilepsy sample reported anti-NMDAR 4%, anti-GAD65 4%, and LGI1 0%.
Interpretation: those numbers argue against indiscriminate antibody-panel enthusiasm. A positive antibody can reclassify a seizure disorder, but a negative panel does not rule out all immune mechanisms, and isolated seropositivity does not prove active autoimmune brain inflammation.
Diagnostic context: autoimmune encephalitis criteria still depend on the whole pattern: subacute clinical course, seizures, mental-status change, MRI, EEG, CSF findings, and antibody results.
Immunotherapy Signals Are Strongest in Antibody-Positive Patients
Kunecki et al. summarized an immunotherapy cohort in which 15 patients with antiepileptic-drug-resistant epilepsy and neural autoantibodies were offered treatment, and 12 agreed. Initial regimens included intravenous methylprednisolone, intravenous immunoglobulin (IVIG), plasma exchange, and rituximab, with some patients later receiving prednisone or prednisone plus azathioprine.
The outcome was clinically meaningful but small-sample: 9 of 12 treated patients had an excellent response, defined as more than 50% seizure reduction. Another 2 had a moderate response of 20-50% seizure reduction, and 1 did not respond.
IVIG is pooled donor immunoglobulin used to modulate immune activity through several possible mechanisms, including neutralizing pathogenic antibodies and altering cytokine signaling. Plasma exchange physically removes antibodies and immune proteins from blood. Rituximab depletes CD20-positive B cells, reducing antibody-producing immune activity upstream. Corticosteroids broadly suppress inflammatory signaling.
These treatments are not benign add-ons. They can carry infection risk, infusion reactions, metabolic effects, blood-pressure effects, psychiatric effects, and cost. The response data therefore support a stepped question: is there enough clinical, antibody, CSF, imaging, EEG, or inflammatory evidence to justify immune-directed treatment in this person?
Children and Adults May Not Share the Same Immune Pattern
Kunecki et al. separated pediatric and adult logic. In children, drug-resistant epilepsy may involve broader inflammatory and developmental mechanisms even when a single defined antibody is not found.
Pediatric immune-marker signal: the review cited ACTH-related immunomodulatory work in children, with immune-marker shifts including increased IL-1β, increased IL-8, increased macrophage inflammatory protein-1 alpha, and decreases in IL-6, IFN-γ, and MCP-1 after treatment.
Adults more often fit a neuronal-surface-antibody model. Anti-NMDAR, LGI1, CASPR2, and related antibodies can disrupt synaptic neurotransmission and often point toward focal seizures, limbic encephalitis, psychiatric symptoms, memory change, sleep disturbance, or autonomic features.
Clinical implication: antibody-negative pediatric cases should not be dismissed automatically if the clinical syndrome is inflammatory, abrupt, severe, or otherwise atypical. In adults, antibody specificity and malignancy screening often carry more weight, especially when onconeural or paraneoplastic antibodies enter the picture.
Paraneoplastic and Encephalitis Workups Belong in the Differential
Autoimmune encephalitis is brain inflammation caused by immune attack, often with seizures, memory impairment, psychiatric symptoms, movement abnormalities, dysautonomia, or altered consciousness. The review placed focal drug-resistant epilepsy beside this larger diagnostic category because seizures can be the presenting feature or a persistent residue of the immune attack.
Paraneoplastic neurologic syndromes are immune-mediated nervous-system disorders triggered by cancer. Antibodies such as anti-Hu, anti-Yo, anti-Ma2, and CRMP5 can signal cytotoxic T cell-mediated neuronal injury and should prompt malignancy search, often with whole-body PET/CT when clinical suspicion is high.
Fluid biomarkers can help size the injury but cannot replace diagnosis. Neurofilament light chain (NfL) reflects axonal injury, while glial fibrillary acidic protein (GFAP) reflects astrocyte activation. Elevated values can support disease-activity assessment, but they do not by themselves prove autoimmune epilepsy.
The Evidence Supports an Immune-Epilepsy Triage Lane
The strongest interpretation is operational. Autoimmune mechanisms should become part of the drug-resistant focal epilepsy workup when ordinary explanations fail or immune features are present.
- Test when the phenotype fits: abrupt onset, new psychiatric or cognitive symptoms, inflammatory CSF, limbic MRI changes, status epilepticus, systemic autoimmunity, or unexplained focal seizures should raise the priority of antibody and autoimmune encephalitis evaluation.
- Treat when the evidence converges: immunotherapy is most defensible when clinical course, biomarkers, antibody results, MRI, EEG, and CSF data point in the same direction.
- Keep surgery and standard epilepsy care active: immune workup should not delay epilepsy surgery evaluation, medication optimization, seizure-safety counseling, or neuromodulation review when those are indicated.
Evidence-strength note: Kunecki et al. synthesized a heterogeneous literature that included case reports, clinical trial registry records, biomarker studies, and narrative evidence. The review did not perform formal risk-of-bias scoring, and the immunotherapy response signal depends heavily on small selected cohorts. The finding is enough to justify better triage; it is not enough to justify broad immunosuppression for all drug-resistant focal epilepsy.
Questions About Autoimmune Drug-Resistant Focal Epilepsy
Should every patient with drug-resistant focal epilepsy get antibody testing?
Not automatically. Testing is most useful when the clinical pattern suggests immune involvement: rapid onset, cognitive or psychiatric change, inflammatory CSF, limbic MRI findings, status epilepticus, autoimmune disease, or otherwise unexplained focal seizures.
Does a positive neural antibody prove autoimmune epilepsy?
No. Antibody results need clinical context because isolated seropositivity can occur without active autoimmune brain inflammation. The diagnosis becomes stronger when antibodies match the seizure phenotype, MRI, EEG, CSF, and clinical course.
When is immunotherapy most defensible?
Immunotherapy is most defensible when drug resistance is paired with immune evidence, especially neuronal surface antibodies, autoimmune encephalitis features, inflammatory CSF, or a subacute syndrome that does not fit ordinary chronic focal epilepsy. The best available response data come from selected antibody-positive patients, not unselected refractory epilepsy clinics.
References
- Kunecki KA, Dziadkowiak E. Autoimmune mechanisms in drug-resistant focal epilepsy: Pathophysiology, biomarkers, and therapeutic implications. Advances in Clinical and Experimental Medicine. 2026. doi:10.17219/acem/218006
- Mesraoua B, Brigo F, Lattanzi S, Abou-Khalil B, Al Hail H, Asadi-Pooya AA. Drug-resistant epilepsy: Definition, pathophysiology, and management. Journal of the Neurological Sciences. 2023;452:120766. doi:10.1016/j.jns.2023.120766
- Iorio R, Assenza G, Tombini M, et al. The detection of neural autoantibodies in patients with antiepileptic-drug-resistant epilepsy predicts response to immunotherapy. European Journal of Neurology. 2015;22:70-78. doi:10.1111/ene.12529
- Dubey D, Alqallaf A, Hays R, et al. Neurological autoantibody prevalence in epilepsy of unknown etiology. JAMA Neurology. 2017;74:397-402. doi:10.1001/jamaneurol.2016.5429
- Cabezudo-Garcia P, Mena-Vazquez N, Ciano-Petersen NL, Garcia-Martin G, Estivill-Torrus G, Serrano-Castro PJ. Prevalence of neural autoantibodies in epilepsy of unknown etiology: Systematic review and meta-analysis. Brain Sciences. 2021;11:392. doi:10.3390/brainsci11030392
- Mohamed H, Hemeda M, Gaber A, et al. Neuronal autoantibodies in a sample of Egyptian patients with drug-resistant epilepsy. Egyptian Journal of Neurology, Psychiatry and Neurosurgery. 2023;59:83. doi:10.1186/s41983-023-00685-9
- Kaczorowska M, Czekuc-Kryskiewicz E, Dadalski M, Kotulska K. Immunological markers of drug resistant epilepsy and its response to immunomodulatory therapy with ACTH in children. Folia Neuropathologica. 2023;61:360-370. doi:10.5114/fn.2023.131662
- Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurology. 2016;15:391-404. doi:10.1016/S1474-4422(15)00401-9