
A 2026 male-mouse study found that 30 mg/kg dexamethasone for 5 days increased light-phase wakefulness (t13 = 4.122, p = 0.0012), decreased NREM sleep (t13 = 4.412, p = 0.0007), and amplified orexin-neuron activity during NREM-to-wake transitions (F1,9 = 6.941, p = 0.0272). The result points to orexin-linked hyperarousal as a testable explanation for steroid-induced insomnia, more than a vague “steroids disrupt sleep” complaint.1
Research Highlights
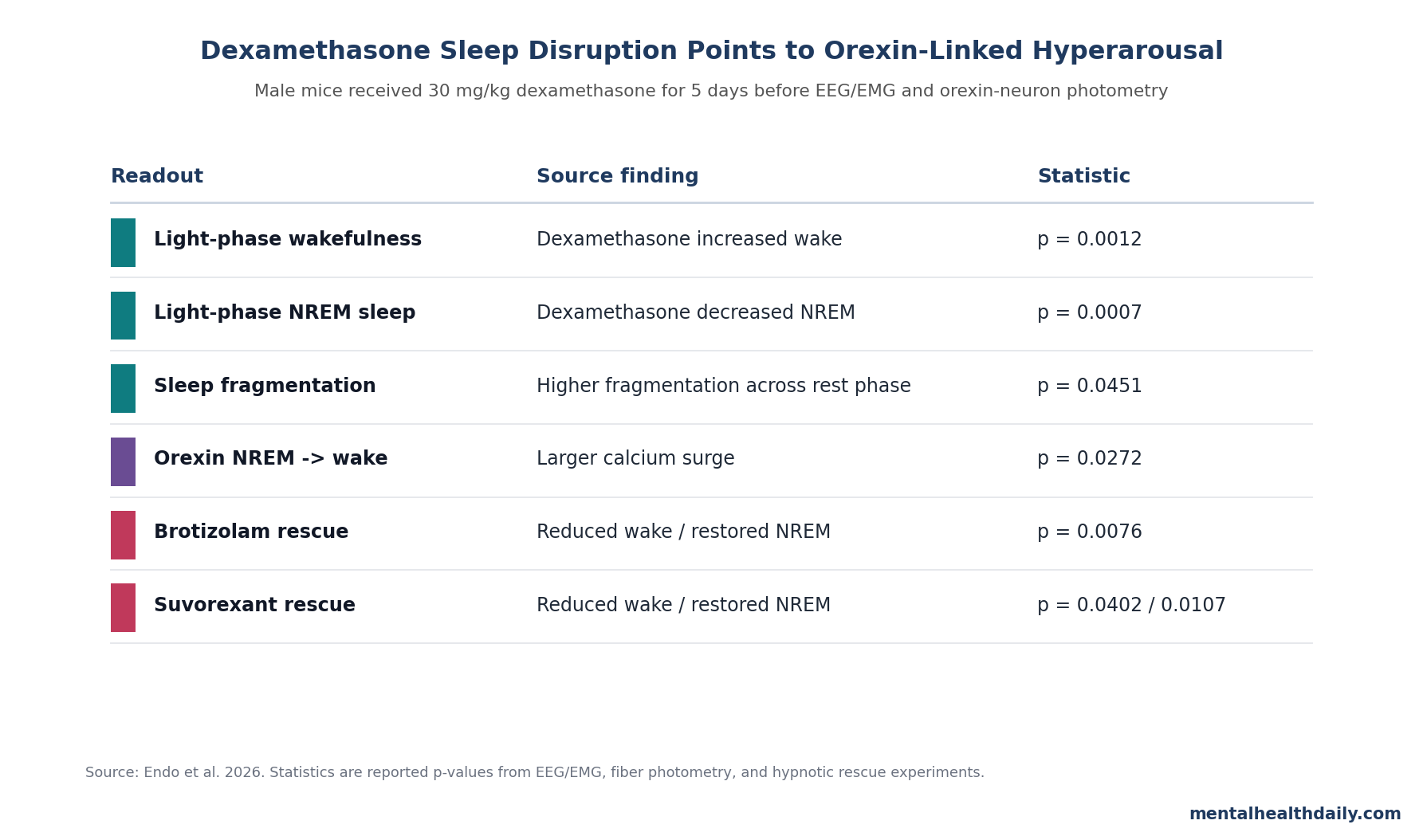
- 5 days of dexamethasone changed the sleep phase: male mice received 30 mg/kg intraperitoneal dexamethasone once daily at ZT 12, then underwent 24 h EEG/EMG recording.1
- Rest-phase sleep was disrupted: during the light phase, dexamethasone increased wakefulness (p = 0.0012), decreased NREM sleep (p = 0.0007), and increased sleep fragmentation across 3-hour bins (F1,14 = 4.842, p = 0.0451).1
- Active-phase sleep moved the other way: during the dark phase, dexamethasone decreased wakefulness (p = 0.0013) and increased NREM sleep (p = 0.0006), a mouse analogue of sleep-wake redistribution rather than simple 24 h arousal.1
- Orexin neurons became overactive at awakening: fiber photometry showed larger orexin-neuron calcium surges than vehicle-treated controls during NREM-to-wake transitions (p = 0.0272; post hoc p = 0.0048) and REM-to-wake transitions (post hoc p = 0.0129).1
- Both hypnotic strategies rescued sleep in mice: 0.3 mg/kg brotizolam and 30 mg/kg suvorexant reduced dexamethasone-linked wakefulness and restored NREM sleep; suvorexant also increased REM duration (p = 0.0124 in Dex-Veh vs. Dex-Suv).1
Dexamethasone is a long-acting synthetic glucocorticoid used for cancer care, autoimmune disease, severe inflammation, cerebral edema, and many other high-stakes indications. The clinical problem is not whether steroids are useful.
They are. The problem is that systemic glucocorticoids can produce insomnia, mood instability, anxiety, delirium, and psychosis, with older clinical reviews estimating mild-to-moderate psychiatric reactions in roughly 28% of patients and severe reactions in nearly 6%.2,3
Endo et al. tested a narrower question: when dexamethasone disrupts sleep, is the arousal system itself overactive, and can different hypnotic classes reverse the phenotype? Their answer was mostly yes in male mice.
The study built a steroid-insomnia model, linked it to orexin-neuron hyperactivity during sleep-to-wake transitions, and showed rescue with both a benzodiazepine receptor agonist and a dual orexin receptor antagonist.
30 mg/kg Dexamethasone for 5 Days Shifted Sleep-Wake Architecture
The researchers administered dexamethasone or saline once daily for 5 consecutive days at ZT 12, the beginning of the dark phase for nocturnal mice. EEG and EMG were recorded for 24 h on the final day.
Sleep was scored in 4-second epochs as wakefulness, non-rapid eye movement sleep (NREM; the deeper non-dream sleep stage), or rapid eye movement sleep (REM; the stage with theta-dominant EEG and muscle atonia).1
The model produced a phase-specific pattern. During the light phase, when mice normally sleep, dexamethasone increased wakefulness (t13 = 4.122, p = 0.0012) and decreased NREM sleep (t13 = 4.412, p = 0.0007).
REM sleep did not significantly change in the main dexamethasone-vs-saline comparison.
During the dark phase, when mice are normally more active, the direction flipped: dexamethasone decreased wakefulness (t13 = 4.095, p = 0.0013) and increased NREM sleep (t13 = 4.538, p = 0.0006). That biphasic pattern is important.
The drug redistributed sleep-wake timing across the full day: more wake during the normal rest phase and more sleep during the normal active phase.
The sleep fragmentation index added a stability readout. Sleep fragmentation index means the number of sleep-to-wake transitions divided by total sleep time, so higher values indicate more broken sleep.
Dexamethasone produced a significant main effect on fragmentation across light-phase 3-hour bins (F1,14 = 4.842, p = 0.0451), while late light-phase NREM sleep was specifically reduced at ZT 9-11 (post hoc p = 0.0241).1
Orexin Neurons Fired More Strongly During Sleep-to-Wake Transitions
Orexin, also called hypocretin, is a hypothalamic neuropeptide system that stabilizes wakefulness. Loss of orexin signaling causes narcolepsy; excessive orexin-linked arousal is a plausible route to insomnia.
Endo et al. tested this mechanism with Orexin-tTA mice expressing GCaMP6, a calcium indicator that allows optical recording of orexin-neuron activity during sleep-wake transitions.1,4
Fiber photometry showed the strongest signal at the moments when mice moved from sleep into wakefulness. During NREM-to-wake transitions, dexamethasone-treated mice had a larger orexin-neuron calcium surge than saline controls (F1,9 = 6.941, p = 0.0272; post hoc p = 0.0048).
During REM-to-wake transitions, the overall comparison was borderline (F1,9 = 5.150, p = 0.0520), but the wakefulness-phase post hoc comparison was significant (p = 0.0129).1
The directionality was also specific. When mice moved from wakefulness into NREM sleep, photometry traces showed no apparent orexin activity in the 10-second window around the transition in either group.
The paper’s interpretation is therefore not that dexamethasone prevented orexin neurons from shutting down for sleep entry. It sensitized the arousal system during awakenings, making sleep more breakable.
Allopregnanolone and Hypothalamic CRH Did Not Explain the Sleep Disruption
The researchers also checked 2 alternative pathways. Allopregnanolone is a neurosteroid that positively modulates GABAA receptors and can influence sleep and sedation.
Plasma allopregnanolone did not differ significantly between saline and dexamethasone groups, and regression analyses did not show significant correlations with wake, NREM, or REM duration across light or dark phases.1
Corticotropin-releasing hormone (CRH) is an upstream hypothalamic stress-system signal that can activate orexin neurons. The study found no significant correlation between hypothalamic CRH concentrations and sleep architecture in treated mice.
These null checks do not rule out every stress-hormone pathway, but they make the orexin-transition signal harder to dismiss as a secondary allopregnanolone or CRH artifact.
Brotizolam and Suvorexant Both Reversed the Mouse Sleep Phenotype
The treatment experiments compared 2 mechanistic strategies after the dexamethasone phenotype had been established. Brotizolam is a benzodiazepine receptor agonist that enhances GABAA-mediated inhibition and produces broad sedation.
Suvorexant is a dual orexin receptor antagonist that blocks orexin-mediated wake-promoting signaling more directly.
After 5 days of dexamethasone or saline pretreatment, the researchers administered 0.3 mg/kg brotizolam or saline at ZT 0, the start of the light phase. Dexamethasone again increased light-phase wakefulness (F1,29 = 20.02, p = 0.0001; Sal-Sal vs. Dex-Sal p = 0.0083) and reduced NREM sleep (F1,29 = 20.23, p = 0.0001; p = 0.0048).
Brotizolam reduced wakefulness in dexamethasone-pretreated mice (Dex-Sal vs. Dex-Bro p = 0.0076) and increased NREM sleep (p = 0.0076), without a significant REM effect.1
Suvorexant produced a similar rescue through the orexin-targeted route. After dexamethasone pretreatment, 30 mg/kg suvorexant decreased wakefulness vs. vehicle (p = 0.0402) and restored NREM sleep (p = 0.0107).
Unlike brotizolam, suvorexant also increased REM duration, with a drug main effect (F1,18 = 14.68, p = 0.0012) and a significant Dex-Veh vs. Dex-Suv comparison (p = 0.0124).1
That does not make suvorexant clinically proven for steroid-induced insomnia. It does make orexin antagonism a rational target for human trials, especially because the same paper directly observed orexin-neuron overactivation during awakenings.
Why This Updates the Steroid-Insomnia Treatment Question
Clinical management of steroid-induced insomnia is often empirical: reduce dose if medically possible, move dosing earlier, try sleep-hygiene measures, and consider a hypnotic when symptoms are severe. That approach is practical, but it is not mechanism-specific.
The Endo study supports a more targeted model:
- Rest-phase insomnia signal: dexamethasone increased wakefulness during the mouse rest phase, the closest analogue to patients being awake at night.
- Transition instability: orexin neurons were most overactive during sleep-to-wake transitions, matching a fragmented-sleep mechanism rather than only delayed sleep onset.
- Orexin-antagonist plausibility: suvorexant reversed the light-phase wake/NREM changes and increased REM sleep in the model.
This also helps explain why a generic sedative answer may be incomplete. Zhao et al. studied 240 cancer patients receiving high-dose glucocorticoid chemotherapy and reported that conventional hypnotics, including benzodiazepines, did not significantly improve subjective sleep quality.5
That clinical result is not a direct contradiction of Endo et al.; it used different endpoints, drugs, patients, and disease context. It does show why steroid-induced insomnia needs direct human trials instead of borrowed insomnia assumptions.
Translation Limits for a High-Dose Male-Mouse Model
The result is useful because it is mechanistic. The result is limited for the same reason: it comes from an intentionally strong animal model.
- 30 mg/kg is a high mouse dose. The researchers selected it to maximize sleep fragmentation and isolate the neural mechanism. Standard human regimens vary widely and cannot be translated by simple body-weight scaling.
- Most C57BL/6 experiments used male mice. Sleep-wake patterns and stress responses differ by sex, so female-mouse and human sex-difference data are still needed.
- Photometry is not causality by itself. Fiber photometry showed orexin-neuron activity rising during awakenings, but it did not selectively silence orexin neurons during dexamethasone exposure. The suvorexant rescue strengthens the case but does not prove every awakening was orexin-driven.
- Hypnotics were tested acutely. Brotizolam and suvorexant were given after the sleep phenotype had developed. Preventive co-administration with steroids, repeated hypnotic dosing, next-day function, delirium risk, and tolerance were not tested.
The honest read: this is strong preclinical evidence for orexin-linked steroid sleep fragmentation, not a patient-level prescribing rule.
Questions About Dexamethasone, Orexin, and Sleep
What did Endo et al. actually show?
The researchers showed that 5 days of 30 mg/kg dexamethasone disrupted male-mouse sleep timing: more wakefulness and less NREM sleep during the light/rest phase, with the opposite shift during the dark/active phase. They also showed larger orexin-neuron calcium surges than vehicle-treated controls during sleep-to-wake transitions and rescue with brotizolam or suvorexant.1
Does this mean suvorexant is better than benzodiazepines for steroid insomnia?
No. In this mouse model, both brotizolam and suvorexant improved the dexamethasone sleep phenotype.
Suvorexant is mechanistically attractive because it targets orexin signaling, but the study did not run a human head-to-head steroid-insomnia trial.
Why is orexin relevant to steroid-induced insomnia?
Orexin neurons stabilize wakefulness. In dexamethasone-treated mice, orexin-neuron activity rose more strongly during NREM-to-wake and REM-to-wake transitions, suggesting the steroid model made the arousal system more reactive at moments when sleep broke into wakefulness.
Did dexamethasone reduce REM sleep?
Not in the main dexamethasone-vs-saline sleep-architecture comparison. The clearest dexamethasone effects were increased wakefulness, decreased NREM sleep during the light phase, altered dark-phase sleep timing, and higher sleep fragmentation.
Suvorexant later increased REM duration in the treatment experiment.1
Can this mouse study guide human steroid dosing time?
Only indirectly. Morning dosing is already used clinically to better align glucocorticoid exposure with the natural cortisol rhythm and reduce sleep disruption when medically feasible.
Endo et al. strengthens the biological rationale for respecting sleep-wake timing, but it did not compare morning vs. evening dosing in humans.
What study would change clinical practice?
A randomized trial in patients starting high-dose glucocorticoids would be the useful test: standard care vs. an orexin antagonist vs. a conventional hypnotic, with polysomnography or wearable sleep architecture, daytime function, delirium, mood, and steroid-treatment adherence measured prospectively.
References
- Endo H, Oue Y, Semba M, et al. Dexamethasone induces sleep disruption in male mice and is associated with hyperactivation of orexin neurons. Psychopharmacology. 2026. doi:10.1007/s00213-026-07057-0
- Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clinic Proceedings. 2006;81(10):1361–1367. doi:10.4065/81.10.1361
- Dubovsky AN, Arvikar S, Stern TA, Axelrod L. The neuropsychiatric complications of glucocorticoid use: steroid psychosis revisited. Psychosomatics. 2012;53(2):103–115. doi:10.1016/j.psym.2011.12.007
- Muehlan C, Roch C, Vaillant C, Dingemanse J. The orexin story and orexin receptor antagonists for the treatment of insomnia. Journal of Sleep Research. 2023;32(6):e13902. doi:10.1111/jsr.13902
- Zhao J, Dai YH, Xi QS, Yu SY. A clinical study on insomnia in patients with cancer during chemotherapy containing high-dose glucocorticoids. Pharmazie. 2013;68(6):421–427. doi:10.1691/ph.2013.2876
- Kato T, Okawa G, Tanaka KF, Mitsukura Y. Dexamethasone induces sleep disturbance in a dose-dependent manner in mice. PLOS ONE. 2023;18(12):e0296028. doi:10.1371/journal.pone.0296028
- Kram DE, Krasnow SM, Levasseur PR, Zhu X, Stork LC, Marks DL. Dexamethasone chemotherapy does not disrupt orexin signaling. PLOS ONE. 2016;11(12):e0168731. doi:10.1371/journal.pone.0168731