
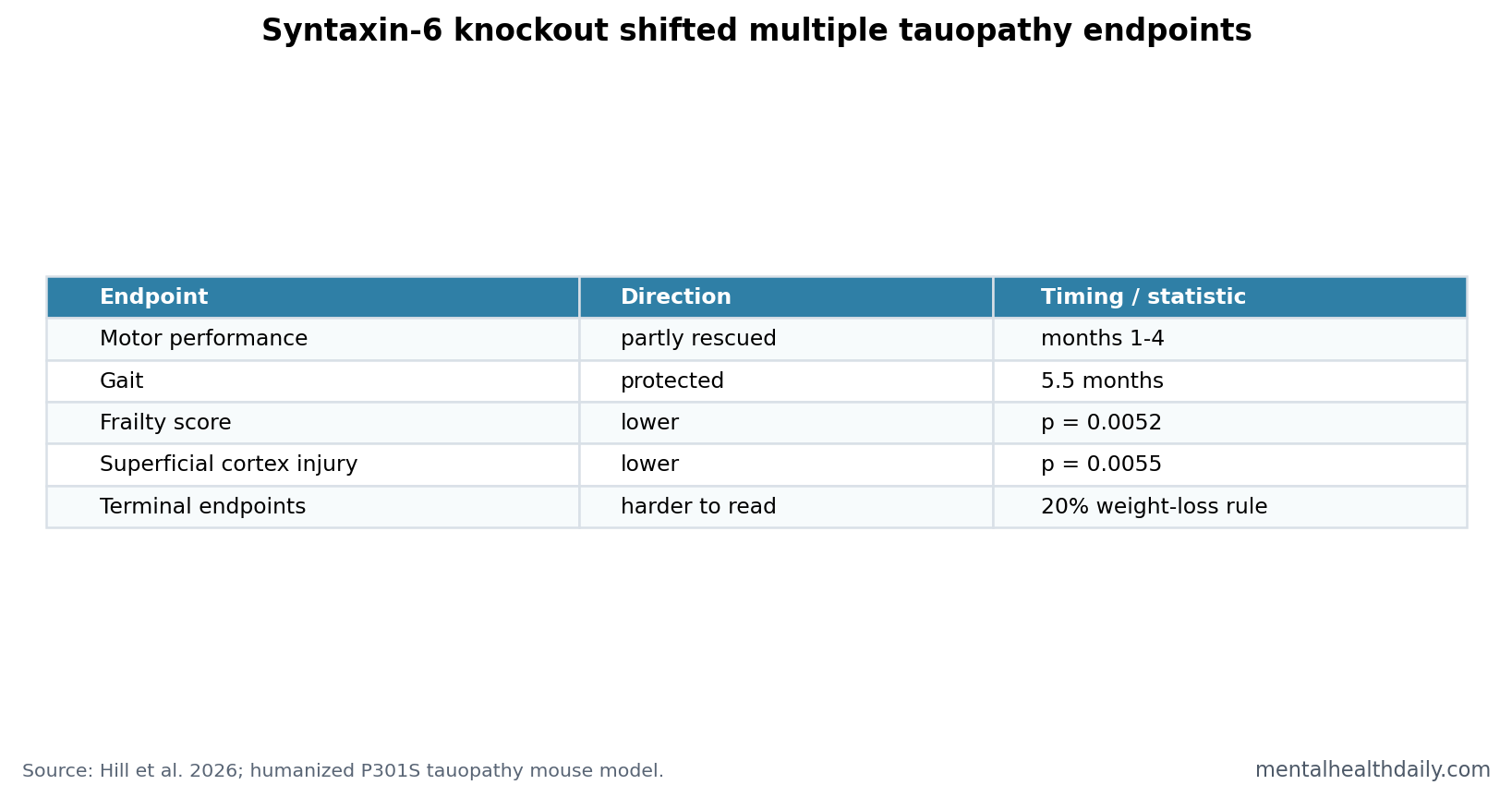
A 2026 humanized P301S tauopathy mouse study found that syntaxin-6 knockout partly rescued early motor impairment from months 1 to 4, protected gait at 5.5 months, reduced frailty (p = 0.0052), and reduced superficial-cortex neurodegeneration at 3 months (p = 0.0055).1 The finding strengthens syntaxin-6 as a tauopathy modifier, but it is still mouse genetics, not a treatment trial.
Research Highlights
- Early motor rescue appeared: P301S tau mice showed motor impairment from 1 month, and syntaxin-6 knockout partly rescued performance from months 1 to 4.1
- Gait was protected later: The knockout group showed additional gait protection at 5.5 months, when disease-relevant gait parameters were already abnormal in tau mice.1
- Frailty moved with genotype: Syntaxin-6 knockout reduced cumulative frailty score in tau mice (p = 0.0052).1
- Cortical injury was partly reduced: Superficial-cortex neurodegeneration at 3 months was lower with syntaxin-6 knockout (p = 0.0055).1
- Translation remains early: A 20% weight-loss culling rule confounded terminal endpoint comparisons, and genetic knockout is not the same as a drug.1
Syntaxin-6 is an intracellular trafficking protein, part of the machinery that helps move cargo through cell compartments. Tauopathy means a neurodegenerative disease process in which abnormal tau protein accumulates and disrupts neurons, as in progressive supranuclear palsy, corticobasal degeneration, some frontotemporal dementia, and Alzheimer’s disease.
Hill et al. asked a functional question: if syntaxin-6 is genetically removed, do early tauopathy phenotypes change in a humanized P301S tau mouse model?
P301S Tau Mice Developed Motor Impairment by 1 Month
The model used humanized P301S tau transgenic mice. The P301S mutation is a tau mutation associated with frontotemporal dementia and parkinsonism in humans, and it produces early motor and neuropathological phenotypes in mice.1
Researchers compared tau mice with normal syntaxin-6 against tau mice lacking syntaxin-6. Rotarod testing captured motor performance, gait analysis captured walking abnormalities, frailty scoring tracked whole-animal health, and tissue analyses examined neurodegeneration and synaptic coverage.
Motor and Frailty Signals Pointed in the Same Direction
The behavioral result was not a single isolated p-value. Tau mice with intact syntaxin-6 developed motor impairment from 1 month of age. Syntaxin-6 knockout partially rescued motor performance from months 1 to 4 and protected gait at 5.5 months.1
Physiology supported the same direction. Knockout mice had more favorable weight trajectories and lower frailty. Frailty differences were statistically clear, with syntaxin-6 knockout reducing cumulative frailty score in tau mice (p = 0.0052).1
Interpretive key: motor rescue, gait protection, weight trajectory, and frailty all point toward a disease-modifying effect in this mouse system. The pattern is stronger than a one-off behavioral anomaly.
Cortical Injury Improved Before End-Stage Disease
Neuropathology gave the study its mechanistic weight. At 3 months, syntaxin-6 knockout reduced neurodegeneration in superficial cortex (p = 0.0055). Later endpoints were more difficult to interpret because of survival and culling effects.1
The paper also reported preserved synaptic coverage at 5 months. Synaptic coverage means how much synapse-associated staining remains around neurons; loss of synaptic coverage is one way neurodegenerative disease can impair circuit function before every neuron is gone.
The terminal caveat is important. Animals were culled at 20% weight loss. Syntaxin-6 knockout mice maintained higher absolute weight, so terminal comparisons were partly shaped by the humane endpoint rule rather than identical disease timing.
Early endpoints carry more weight in this design because they were measured before that terminal-selection problem dominated interpretation. The month 1 to month 4 motor rescue, 5.5-month gait protection, frailty difference, and 3-month superficial-cortex result all sit closer to the period where genotype effects can be read with fewer end-stage complications.
Mechanistic reading: syntaxin-6 appears to modify how tau pathology becomes organism-level dysfunction. That does not require every pathology marker to move in parallel. A modifier can improve motor function, synaptic preservation, or tissue vulnerability while leaving some tau biochemical measures less changed.
Syntaxin Biology Fits a Wider Tau-Trafficking Story
Syntaxin-6 did not enter this paper as a random gene. Prior work linked syntaxin biology to disease-risk and protein-trafficking questions. Hill et al. previously examined syntaxin-6 in prion cellular phenotypes, while Lee et al. reported that syntaxins 6 and 8 can facilitate tau entry into secretory pathways.23
That makes the tauopathy mouse result more coherent. Tau disease includes how much tau exists, where tau travels, how it seeds pathology, how cells handle misfolded protein, and how vulnerable circuits respond.
Human tau-lowering work adds the therapeutic context. Mummery et al. tested a tau-targeting antisense oligonucleotide in mild Alzheimer’s disease, showing that tau biology can be targeted in humans, even though syntaxin-6 itself has not been validated as a human therapy.4
Knockout Benefit Is Not Automatically a Drug Strategy
Genetic knockout is a blunt tool. A mouse born without syntaxin-6 is not the same as an older adult taking a selective syntaxin-6 modulator after tauopathy symptoms begin. A drug would need timing, dose, tissue specificity, safety, and evidence that modifying syntaxin-6 does not disrupt necessary trafficking in other cells.
That translation problem has 3 layers:
- Timing: the mouse model changes syntaxin-6 from development onward, while human treatment would usually begin after pathology has already accumulated.
- Specificity: syntaxin-6 participates in intracellular trafficking, so broad inhibition could affect healthy cells as well as vulnerable tau-burdened neurons.
- Disease match: a P301S mouse model captures parts of human tauopathy but cannot represent the full mixture of aging, vascular disease, inflammation, amyloid, and comorbidity seen in patients.
The strongest near-term use is target validation. A functional genetic modifier that improves motor, gait, frailty, synaptic, and cortical endpoints gives researchers a better reason to map syntaxin-6 pathways in human tissue, organoid systems, and drug screens.
Clinical boundary: the result should not be read as a reason to change care for Alzheimer’s disease, progressive supranuclear palsy, or frontotemporal dementia today. It is a mechanistic clue that may help prioritize drug-discovery work.
Drug discovery would also need directionality. Full knockout may be protective in this model, but partial inhibition, timing-limited inhibition, or pathway-specific modulation could behave differently. Syntaxin-6 may also have different roles across neurons, glia, and peripheral tissues.
Human genetics can help decide whether a target is worth pursuing. Genes supported by human disease association tend to have better odds in drug development, but mouse functional validation remains necessary when the human signal is indirect. This paper strengthens the functional side of the argument.
Evidence-strength note: this was an animal-model mechanism study. It can validate syntaxin-6 as a functional modifier in a P301S tau model. It cannot show that syntaxin-6 inhibition treats human Alzheimer’s disease, progressive supranuclear palsy, corticobasal degeneration, or frontotemporal dementia.
Human Tauopathy Translation Needs Disease-Specific Testing
Tauopathies are not one disease. Progressive supranuclear palsy, corticobasal degeneration, frontotemporal dementia with tau mutations, and Alzheimer’s disease differ in anatomy, age of onset, co-pathology, and clinical presentation. A modifier that helps one model may be weaker in another.
A useful translational path would test syntaxin-6 manipulation across several tau models, then compare the direction of effect with human tissue and genetic data. Consistent direction across models would make the target more credible. Divergent direction would tell researchers where the biology is disease-specific.
For readers, the current result is best read as a target-prioritization paper. It raises syntaxin-6 above background noise in tauopathy biology, while leaving the treatment question open.
The practical research standard is replication across function and pathology. If syntaxin-6 modulation improves movement but worsens another neural process, the target becomes harder. If it repeatedly protects behavior, synapses, and vulnerable cortical regions, the drug-development case becomes cleaner.
That balance is especially important for trafficking proteins. The same transport machinery that could reduce tau-related injury may also support normal cellular housekeeping. Therapeutic interest therefore depends on finding a controllable pathway window with tissue, dose, and timing constraints.
For now, syntaxin-6 is a sharper biological lead than a clinical lever, and the next experiments should treat safety, reversibility, timing, and dosing as part of target validation.
Therapeutic test: conditional or adult-onset syntaxin-6 reduction would be more informative than lifelong knockout alone. If benefit persists when syntaxin-6 is lowered after development and after early pathology has started, the result would look more like a druggable intervention and less like a developmental compensation effect.
- Timing test: lower syntaxin-6 after tau pathology starts.
- Safety test: measure trafficking, synapses, weight, and peripheral toxicity.
- Disease test: repeat across tauopathy models with different anatomy.
Those tests would decide whether syntaxin-6 is a durable target or a model-specific modifier.
The answer determines whether drug discovery should keep moving.
The current data justify that work.
Questions About Syntaxin-6 and Tauopathy
Did the study test a syntaxin-6 drug?
No. It tested genetic knockout in mice, not a medication.
Does this apply to Alzheimer’s disease?
Only indirectly. Alzheimer’s disease includes tau pathology, but this study used a P301S tauopathy mouse model and did not test patients.
Why does the 20% weight-loss rule matter?
It means terminal comparisons were partly influenced by when animals reached a humane endpoint. That makes late-stage readouts harder to interpret than early motor, gait, frailty, and cortical findings.
References
- Hill E, et al. Modification of early behavioural, physiological and neuropathological endpoints by syntaxin-6 knockout in a humanised P301S transgenic model of tauopathy. Acta Neuropathologica. 2026. https://doi.org/10.1007/s00401-026-03009-2
- Hill E, et al. Intracellular trafficking SNARE protein, syntaxin-6, modifies prion cellular phenotypes and risk of disease development in vivo. Acta Neuropathologica. 2025. PubMed search
- Lee WS, et al. Syntaxins 6 and 8 facilitate tau into secretory pathways. Biochemical Journal. 2021. PubMed search
- Mummery CJ, et al. Tau-targeting antisense oligonucleotide MAPTRx in mild Alzheimer’s disease: a phase 1b randomized placebo-controlled trial. Nature Medicine. 2023. PubMed search