
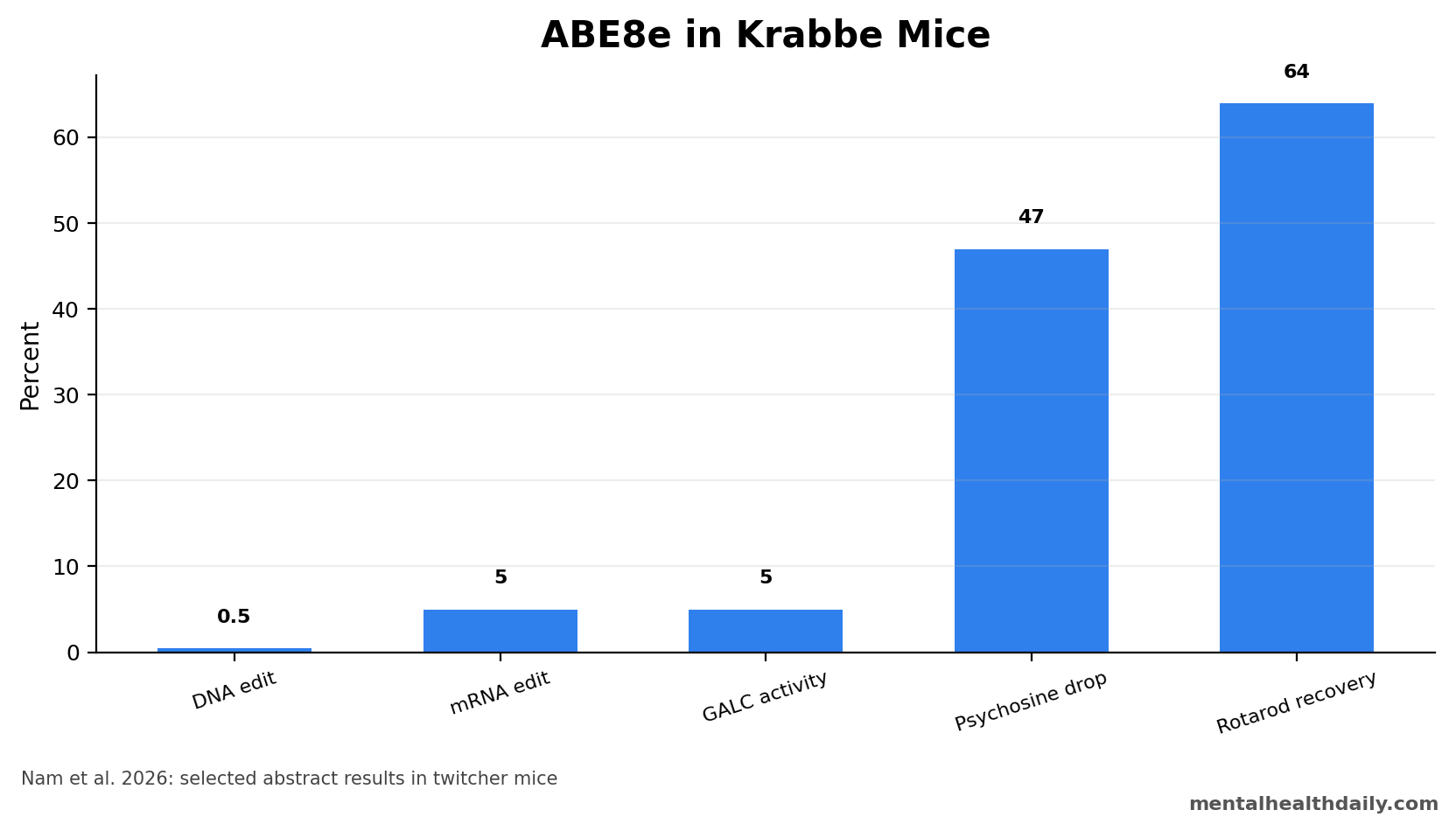
A 2026 twitcher-mouse study found that adenine base editing corrected only about 0.5% of genomic DNA and 5% of mRNA in a Krabbe disease model, yet that low editing signal restored GALC enzyme activity to about 5% of wild-type levels and reduced psychosine by about 47%.1 This mouse experiment shows why even small correction fractions can matter in enzyme-deficiency brain diseases.
Research Highlights
- Editing was low but measurable: ABE8e corrected approximately 0.5% of genomic DNA and 5% of mRNA 3 weeks after dual-AAV9 injection.1
- Biochemistry moved: GALC activity recovered to about 5% of wild-type levels, while psychosine accumulation fell by about 47%.1
- Motor outcomes improved: 5 weeks after treatment, body weight reached about 64% of wild-type, clasping scores about 23%, and rotarod performance about 64%.1
- Myelin was more preserved than in untreated twitcher mice: histology, diffusion tensor imaging, T2-weighted MRI, and electron microscopy all supported better myelination and axonal integrity after ABE8e.1
- Translation is early: 0 human patients were treated, and brain-wide AAV base editing still needs safety and durability testing.1
Krabbe disease, also called globoid cell leukodystrophy, is a lysosomal storage disease caused by pathogenic variants in GALC, the gene encoding galactosylceramidase. GALC normally helps break down psychosine, a toxic lipid that damages oligodendrocytes, the cells that make myelin.
Adenine base editors are CRISPR-derived tools that convert one DNA base to another without making the double-strand break used by standard Cas9 editing. In this study, the target was a premature stop codon in the twitcher mouse Galc gene.
0.5% DNA Correction Was Enough to Move Disease Biology
Nam et al. screened 3 adenine base editor variants in cells and then delivered split ABE8e through dual adeno-associated virus serotype 9 vectors into newborn twitcher mice by intracerebroventricular injection. AAV9 is a viral delivery vehicle often used in nervous-system gene therapy because it can reach neural tissue.
The headline result looks numerically small. Three weeks after injection, correction reached about 0.5% of genomic DNA and 5% of mRNA. Enzyme activity still rose to about 5% of wild-type levels.1
Enzyme biology explains the leverage: lysosomal storage diseases can sometimes improve when a small amount of missing enzyme is restored, because the biochemical bottleneck is not an all-or-none process.
Psychosine Fell by 47% After ABE8e Treatment
Psychosine is the major neurotoxic metabolite in Krabbe disease. It accumulates when GALC activity is deficient and contributes to oligodendrocyte death, demyelination, and neurodegeneration. In Nam et al., ABE8e treatment reduced psychosine by about 47% while improving myelin measures.1
Multiple readouts pointed in the same biological direction:
- Enzyme activity: GALC activity recovered to about 5% of wild-type levels.
- Toxic metabolite: psychosine accumulation dropped by about 47%.
- White matter: MRI, diffusion tensor imaging, histology, and electron microscopy supported better myelination.
- Function: rotarod performance and clasping scores improved, and lifespan extended.
Newborn Brain Delivery Was Central to the Mouse Result
Nam et al. treated twitcher mice on postnatal day 1 by intracerebroventricular injection, meaning the vectors were delivered into the brain’s ventricular fluid space shortly after birth.1 That timing matters. Infantile Krabbe disease progresses quickly, and the mouse experiment targeted the nervous system before later-stage demyelination could dominate the phenotype.
Dual-AAV9 delivery was the engineering workaround. ABE8e is too large for a single adeno-associated virus package, so the researchers split the editor across 2 AAV9 vectors that had to reach the same cells and reconstitute functional editing machinery.1
That design makes the result more technically impressive, but also highlights the translational burden: a human treatment would need reliable distribution, cell-type reach, immune tolerability, and manufacturing control for a split-vector system.
The study also explains why the researchers chose base editing instead of ordinary CRISPR cutting. Base editing changes a target base without creating the double-strand DNA break associated with standard Cas9 editing. For a fragile newborn-brain use case, avoiding double-strand breaks is an obvious safety advantage, even though base editors still require off-target DNA and RNA editing surveillance.
Existing Krabbe Treatments Set a High Bar for Gene Editing
Krabbe disease already has one partially effective clinical path: hematopoietic stem cell transplantation can slow progression when performed very early, especially before symptoms are obvious.4 It is not a clean cure, and it does not repair the GALC mutation, but it gives any future gene-editing strategy a real comparator.
That comparator matters for trial design. A future base-editing program would need to show both biochemical movement and an acceptable risk-benefit profile against early transplant, supportive care, and any emerging gene-transfer approaches. For newborns flagged by screening, a therapy with permanent editing risk has to earn its place with durable function and human-relevant safety, not mouse histology alone.
Why the 5% enzyme signal still matters: lysosomal enzyme disorders can respond to small amounts of restored enzyme because some enzyme can be secreted, taken up by neighboring cells, or enough to reduce toxic substrate pressure. The Nam et al. result fits that biology: low editing, low enzyme restoration, but a larger downstream psychosine reduction and visible motor improvement.1
The result should also be read against the severity of the untreated twitcher model. These mice lose myelin, develop motor dysfunction, and die early, so partial biochemical rescue is meaningful when it appears alongside MRI, diffusion tensor imaging, histology, electron microscopy, rotarod, clasping, body-weight, and lifespan signals. The key interpretation is that a small upstream edit produced several downstream movements in the same direction.
What remains unresolved: the paper does not prove the minimum editing threshold needed for durable benefit, the best delivery route for a human newborn, or whether the same editor would remain active and safe over longer follow-up. Those questions become harder, not easier, as the target moves from a controlled mouse model to infants with different mutations and clinical timelines.
One reason the low editing rate can still move biology is that the target enzyme sits upstream of a toxic-metabolite problem. Psychosine accumulation injures oligodendrocytes, the myelin-making cells that support fast nerve signaling. Reducing psychosine pressure by about 47% therefore gives the nervous system a plausible path to better myelin preservation, even when the original DNA correction fraction looks small.1
The same logic also limits overinterpretation. A 47% psychosine reduction is large for a mouse biochemical marker, but it still leaves residual disease biology. The motor and body-weight results improved toward wild-type values rather than reaching normal levels, which is why the result reads as partial rescue instead of disease elimination.
Mouse Improvement Does Not Equal a Human Treatment
Evidence-strength note: this was an animal gene-editing study. It can show feasibility, biological plausibility, and a rough therapeutic direction. It cannot establish dosing, safety, immune risk, off-target editing risk, or benefit in infants with Krabbe disease.
The delivery problem is especially important. Dual-AAV delivery lets a large base editor be split across 2 viral vectors, but each extra vector adds complexity. Human brain delivery would also need to solve dose, distribution, immune response, durability, and manufacturing questions.
Existing Krabbe disease care has a narrow window. Hematopoietic stem cell transplantation can slow disease when performed very early, yet it does not directly repair the mutant GALC gene. A successful editing strategy would need to beat that risk-benefit standard with durable animal rescue, human-relevant safety, and a realistic newborn treatment path.
Most important limitation: the twitcher mouse model is useful because it carries a defined Galc stop-codon mutation, but human Krabbe disease includes different GALC variants, different diagnostic timing, and a different treatment-delivery problem. A mutation-specific editor that works in one mouse genotype does not automatically generalize to the full human disease population.
Longer follow-up is also essential because early motor improvement can overstate durability in rapidly progressive animal models. A credible translational package would need sustained enzyme activity, stable psychosine reduction, preserved myelin, and no late toxicity after the editing machinery has done its work.
Questions About Krabbe Disease Base Editing
Was 0.5% DNA correction too low to matter?
In this mouse study, no. The low genomic correction was accompanied by about 5% mRNA correction, about 5% GALC activity, a 47% psychosine reduction, and measurable motor improvement.
Did the mice become normal?
No. Body weight and rotarod performance reached about 64% of wild-type levels at 5 weeks, which signals improvement rather than normalization.
What should come next?
The next development step is safety-heavy: broader off-target analysis, durability testing, delivery optimization, and replication in models that better approximate human treatment timing.
Would this apply to every child with Krabbe disease?
No. This editor targeted the twitcher mouse Galc premature stop codon. Human therapy would need mutation matching or a different editing strategy for other GALC variants, plus newborn screening and delivery timing that make treatment early enough to preserve nervous-system function.
Why use adenine base editing instead of standard CRISPR-Cas9?
The appeal of adenine base editing is that it can change the target base without making a double-strand DNA break. That does not make it risk-free, but it is a better conceptual fit for nervous-system repair than a strategy built around cutting DNA in newborn brain cells.
References
- Nam BG, Seo JH, Hong SA, et al. In vivo adenine base editing of mutant Galc gene ameliorates Krabbe disease progression. Genome Medicine. 2026;18:46. https://doi.org/10.1186/s13073-026-01620-2
- Rafi MA, Rao HZ, Luzi P, Wenger DA. Long-term improvements in lifespan and pathology in CNS and PNS after BMT plus one intravenous injection of AAVrh10-GALC in twitcher mice. Molecular Therapy. 2015;23:1681–1690. https://doi.org/10.1038/mt.2015.145
- Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017. https://doi.org/10.1038/nature24644
- Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe disease. New England Journal of Medicine. 2005. https://doi.org/10.1056/nejmoa042604