
A 2026 Nature study found that parasite-sensing tuft cells can release acetylcholine, activate crypt enterochromaffin cells through muscarinic receptors, trigger serotonin release, stimulate vagal afferent neurons, and suppress food intake during established infection.1 The finding is a precise gut-defense circuit, not a generic claim that the gut-brain axis explains every mood or appetite problem.
Research Highlights
- Acetylcholine activated serotonin cells: 10 μM ACh stimulated enterochromaffin-cell calcium signals and serotonin release, with biosensor currents separating EC from non-EC cells (P = 0.0099; n = 7).
- Muscarinic receptors carried the signal: atropine blocked ACh-evoked EC-cell responses (P < 0.0001), while the nicotinic antagonist mecamylamine did not (P = 0.6197).
- Crypt cells were the target: ACh triggered serotonin release in crypts but not villi, with a crypt-villus difference at P < 0.0001 across 17 crypts and 9 villi.
- Two ACh phases explained symptoms: acute parasite-metabolite release came first, while sustained leak-like release during type 2 inflammation produced enough serotonin to activate vagal signaling.
- Behavioral output was protective: vagal afferent activation suppressed food intake during the 2026 established-infection experiments, linking epithelial parasite sensing to brain-directed behavior.
Tuft cells are rare sensory epithelial cells in the gut that detect parasites and help launch type 2 immune defense. Enterochromaffin cells are gut epithelial cells that release serotonin and communicate with sensory nerve fibers, especially when the gut detects irritants, mechanical stress, or injury.
Vagal afferent neurons carry sensory information from the gut to the brain through the vagus nerve. In this study, the gut signal was not vague background inflammation. It was a stepwise epithelial-to-neural pathway.
10 uM Acetylcholine Activated Crypt Serotonin Cells
Touhara et al. first asked whether acetylcholine from tuft cells could activate enterochromaffin cells. In organoid and dissociated-cell experiments, 10 μM acetylcholine robustly stimulated EC-cell calcium responses, comparable to mustard-oil TRPA1 activation or high-potassium depolarization.1
The researchers then used HEK293FT cells expressing serotonin-gated 5-HT3 receptors as biosensors next to EC cells. After acetylcholine stimulation, adjacent biosensor cells showed significant inward currents, indicating serotonin release at levels capable of activating 5-HT3 receptors. EC-cell stimulation separated from non-EC cells with P = 0.0099.
Why this matters: the experiment showed functional chemical handoff. Tuft-cell transmitter could make serotonin-producing epithelial cells send a neural signal.
Atropine Blockade Put Muscarinic Receptors in the Middle
The receptor result was clean. RNA-sequencing pointed to Chrm3 expression in crypt-residing EC cells, and pharmacology supported a muscarinic receptor mechanism. Atropine, a muscarinic antagonist, reduced the ACh-evoked EC response (P < 0.0001). Mecamylamine, a nicotinic antagonist, did not meaningfully block it (P = 0.6197).
Calcium-store depletion also weakened the response, while removing extracellular calcium did not. That pattern fits metabotropic muscarinic signaling, where receptor activation mobilizes intracellular calcium stores rather than opening a fast ion channel at the membrane.
Translation: acetylcholine was not acting as a generic irritant. It used a specific receptor class on a specific epithelial target.
Crypt-Villus Location Determined Serotonin Release
Gut epithelium is spatially organized. Crypts are recessed gland-like zones, while villi project into the intestinal lumen. Touhara et al. found that acetylcholine elicited serotonin release from crypt EC cells, not villus EC cells. The crypt-villus difference reached P < 0.0001 across 17 crypts and 9 villi.
That location detail prevents a sloppy gut-brain-axis story. The pathway was not “parasites make the gut release serotonin everywhere.” The signal depended on where the relevant EC cells sat and which receptor program they expressed.
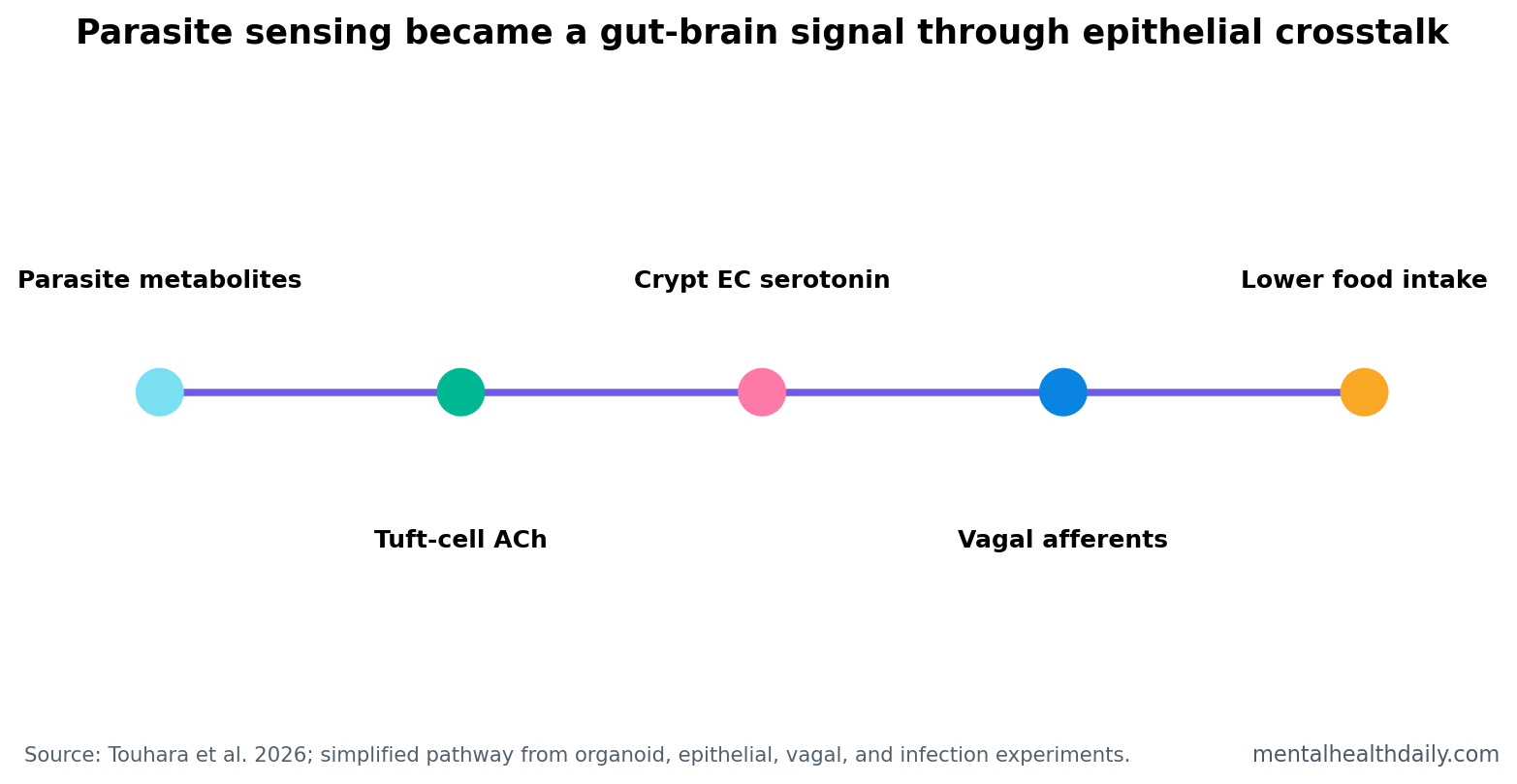
Two Acetylcholine Phases Explained the Shift From Silent to Symptomatic Infection
The study described 2 acetylcholine-release modes. The first was acute release in response to parasite-derived metabolites. The second was sustained, leak-like acetylcholine release during type 2 inflammation.
Both modes could activate muscarinic receptors on crypt EC cells, but only sustained release produced enough serotonin to activate vagal afferent neurons and suppress food intake. That timing explains why parasite detection can begin before the animal shows the full behavioral output of established disease.
Protective-behavior logic: lower food intake during infection may reduce exposure, alter gut conditions, or redirect energy into immune defense. It is not automatically pathological appetite loss; in the experimental context, it was part of host defense.
Gut-Brain Signaling Needs Cell-Level Specificity
The phrase “gut-brain axis” gets overused in wellness and psychiatry writing. This paper is useful because it replaces vague gut-brain branding with cell types, transmitters, receptors, anatomical location, sensory neurons, and behavior.
Cell type: tuft cells and enterochromaffin cells.
Transmitters: acetylcholine from tuft cells and serotonin from EC cells.
Receptor path: muscarinic receptors on crypt EC cells and 5-HT3 receptor-linked sensory signaling.
Behavior: suppressed food intake during established parasite infection.
That level of specificity is the standard for translating gut-brain findings into psychiatry or neurology. A gut signal becomes clinically meaningful only when the pathway and context are clear.
Evidence Strength: Strong Circuit Biology, No Human Treatment Claim
Evidence-strength note: this was a mechanistic study using mouse models, organoids, biosensors, epithelial imaging, pharmacology, and infection experiments. It was not a human trial and did not test depression, anxiety, eating disorders, or irritable bowel syndrome treatment.
The result can inform mental-health science indirectly because serotonin, interoception, vagal signaling, and appetite are all relevant to brain-body regulation. But importing the finding into clinical psychiatry requires caution. Parasite-defense anorexia is not the same as depression-related appetite loss, anorexia nervosa, medication nausea, or functional gastrointestinal symptoms.
Useful translation starts with mechanism. This study says that epithelial immune-sensory cells can convert infection detection into serotonin-mediated vagal signaling and behavior. It does not say that boosting or blocking this pathway is safe or useful in humans.
Why Appetite Suppression Can Be a Defense Signal
Appetite loss is usually treated as a symptom to reverse, and in many clinical settings that is correct. But infection biology is different. During some infections, eating less can be part of a coordinated defense program that changes gut exposure, energy allocation, immune activity, and pathogen environment.
Two-phase logic: the acute acetylcholine release detected parasite-derived metabolites, but sustained leak-like release during type 2 inflammation produced the serotonin level needed to engage vagal afferents. That timing makes the behavioral output conditional on established disease rather than every brief parasite signal.
Why serotonin location matters: gut serotonin is not the same as brain serotonin. Enterochromaffin-cell serotonin can activate local and vagal sensory pathways without implying that central mood circuits are being directly flooded with serotonin.
Why this belongs in neuroscience: the circuit turns an epithelial immune event into a sensory-neural message and then into behavior. That is brain-body regulation at cellular resolution, not a loose metaphor about the gut influencing the mind.
Clinical Translation Should Stay Narrow
The tempting overreach is to turn the result into a serotonin or vagus-nerve story for every appetite symptom. The paper does not support that. It studied parasite-triggered epithelial crosstalk in experimental systems, not psychiatric appetite change in humans.
What it can inform: nausea, infection-related anorexia, visceral hypersensitivity, and inflammatory gut states may involve epithelial sensory cells more than older models assumed. The study gives researchers a concrete circuit to test in related disease contexts.
What it cannot answer: it does not show whether SSRIs, 5-HT3 antagonists, vagus-nerve stimulation, probiotics, antiparasitic therapy, or dietary changes alter this pathway in people with mental-health symptoms. Those would be separate experiments.
Reader-side use: the main takeaway is conceptual. Gut-brain signaling can be highly specific: a pathogen cue, a tuft cell, acetylcholine, a crypt EC cell, serotonin, a vagal afferent, and a behavior. That specificity is the antidote to vague gut-brain claims.
The useful clinical path would start with infection, nausea, and inflammatory gut states where the same epithelial players are plausibly active. Psychiatric translation should come later, after human studies show whether comparable tuft-cell, enterochromaffin-cell, and vagal signatures appear outside the parasite model.
Animal appetite data should also be paired with sickness-behavior measures so reduced feeding is not overread as a single-purpose gut signal.
Why the Receptor Details Matter
Receptor specificity is what turns a pathway into a testable model. If atropine blocks the ACh response while mecamylamine does not, the pathway points toward muscarinic receptor signaling rather than nicotinic receptor signaling. If crypt EC cells respond while villus EC cells do not, the pathway points toward location-specific epithelial programming.
Drug-development implication: a future therapy would have to consider receptor subtype, epithelial location, infection stage, and serotonin output. A blunt serotonin intervention could miss the upstream trigger or create off-target gut effects.
Research implication: related gut-brain studies should name the cell type and receptor rather than stopping at “inflammation increased serotonin.” Without that resolution, appetite, nausea, pain, and mood-related claims become impossible to compare across studies.
Questions About Parasites and Gut-Brain Serotonin
Did the study show that parasites directly affect the brain?
It showed a gut-to-brain signaling route. Parasite-triggered epithelial crosstalk released serotonin, activated vagal afferent neurons, and changed food intake.
Is this about mood serotonin?
Not directly. The serotonin here came from gut enterochromaffin cells and acted on sensory pathways. That is different from central serotonin signaling targeted by antidepressants.
Does this suggest a treatment for appetite or anxiety?
No. It identifies a parasite-defense circuit. Human clinical uses would need separate safety and efficacy studies.
References
- Touhara KK, Xu J, Castro J, et al. Parasites trigger epithelial cell crosstalk to drive gut-brain signaling. Nature. 2026. https://doi.org/10.1038/s41586-026-10281-5
- PubMed search: enterochromaffin cells serotonin vagal afferent 5-HT3. https://pubmed.ncbi.nlm.nih.gov/?term=enterochromaffin+cells+serotonin+vagal+afferent+5-HT3
- PubMed search: tuft cells parasites acetylcholine type 2 immunity. https://pubmed.ncbi.nlm.nih.gov/?term=tuft+cells+parasites+acetylcholine+type+2+immunity
- PubMed search: gut brain axis serotonin food intake vagus nerve. https://pubmed.ncbi.nlm.nih.gov/?term=gut+brain+axis+serotonin+food+intake+vagus+nerve