
A 2026 two-cohort connectome study found that post-stroke depressive symptoms tracked damage to networks weighted by the serotonin transporter and vesicular acetylcholine transporter: 5-HTT damage predicted depression in Leipzig (OR 2.4, 95% CI 1.15–5.02) and Oxford (OR 2.05, 95% CI 1.03–4.09), while VAChT damage also replicated across both cohorts.1 The dopamine part of the original hypothesis did not replicate, which makes this a serotonin-acetylcholine network finding rather than a broad monoamine story.
Research Highlights
- 5-HTT damage replicated: serotonin transporter-weighted network damage predicted post-stroke depressive symptoms in Leipzig (OR 2.4, p = 0.020) and Oxford (OR 2.05, p = 0.041), with both models improving beyond age, sex, lesion volume, and neurological deficit.1
- VAChT damage was the other stable signal: vesicular acetylcholine transporter-weighted network damage predicted depression in Leipzig (OR 3.14, p = 0.042) and Oxford (OR 4.18, p = 0.012), pointing to a cholinergic pathway that standard post-stroke depression summaries often underplay.1
- Dopamine was mixed, not confirmed: D1-weighted network damage was significant in Leipzig (OR 3.63, p = 0.012) but not Oxford (OR 1.74, p = 0.306), opposite the cleaner dopamine prediction a reader might expect from depression-anhedonia models.1
- The analysis used 435 complete cases: 267 Leipzig patients and 168 Oxford patients had lesion, clinical, disability, and 6-month HADS-D data; 97 of 435 met the study threshold for post-stroke depressive symptoms.1
- The result is mechanistic, not diagnostic: Frey et al. used normative PET neurotransmitter maps and population structural connectomes, so the ORs identify systems-level vulnerability rather than a patient-ready prediction test.1
Post-stroke depressive symptoms are depressive symptoms after stroke severe enough to affect recovery, cognition, function, or quality of life. Systematic reviews usually place clinically important depression after stroke near one-third of survivors, but lesion location alone has never explained why 2 people with superficially similar strokes can diverge so sharply in mood outcome.2
Frey et al. tested a more specific claim: the risk may depend on which neurotransmitter-weighted networks a stroke disrupts. A neurotransmitter-weighted connectome combines 2 maps: the brain’s structural wiring and positron-emission tomography (PET) maps showing where receptors or transporters for serotonin, dopamine, acetylcholine, and other systems are most dense.
The lesion is then scored by how much it interrupts wiring embedded in each neurochemical architecture.1
435 Stroke Patients, 19 Neurotransmitter Maps, 2 Replication Cohorts
Researchers reanalyzed Leipzig and Oxford stroke datasets with acute lesion masks and 6-month depression follow-up. The final complete-case sample included 435 patients: 267 from Leipzig and 168 from Oxford.
Post-stroke depressive symptoms were defined as a Hospital Anxiety and Depression Scale depression subscale (HADS-D) score above 7 at follow-up.1
HADS-D is a symptom scale, not a structured psychiatric interview. That distinction matters for interpretation: the study modeled elevated depressive symptoms after stroke, not clinician-confirmed major depressive disorder.
The method had 2 steps. First, partial least squares regression identified neurotransmitter-informed damage scores with variable importance in projection (VIP) above 1.
Second, candidate systems were entered into logistic regression models adjusted for age, sex, lesion volume, National Institutes of Health Stroke Scale (NIHSS) score, and Barthel Index. Model fit was judged against covariate-only models with Delta AIC greater than 2 treated as meaningful improvement.1
This design is useful because it asks a different question from ordinary lesion-volume adjustment. A large lesion can miss the serotonin- or acetylcholine-weighted pathways most relevant to mood symptoms, while a smaller lesion can interrupt a high-value network segment.
The 10% damage increments in Frey et al. are therefore not simple “more brain injury” measures; they are system-weighted disconnection scores.
The 2-cohort structure also keeps the result from resting on one local dataset. Leipzig was the discovery-style cohort, Oxford was the replication-style cohort, and the most important signal is the pattern that survived both settings.
That is why the dopamine result stays secondary even though the Leipzig OR looked large: a neurotransmitter association that fails in Oxford is a lead, not a stable post-stroke depression marker.
Serotonin and Acetylcholine Replicated; D1 Dopamine Did Not
The stated hypothesis emphasized neurotransmitter-informed disruption, “particularly within serotonergic and dopaminergic systems.” The final pattern was mixed: serotonin replicated, dopamine did not, and acetylcholine became the second cross-cohort signal.1
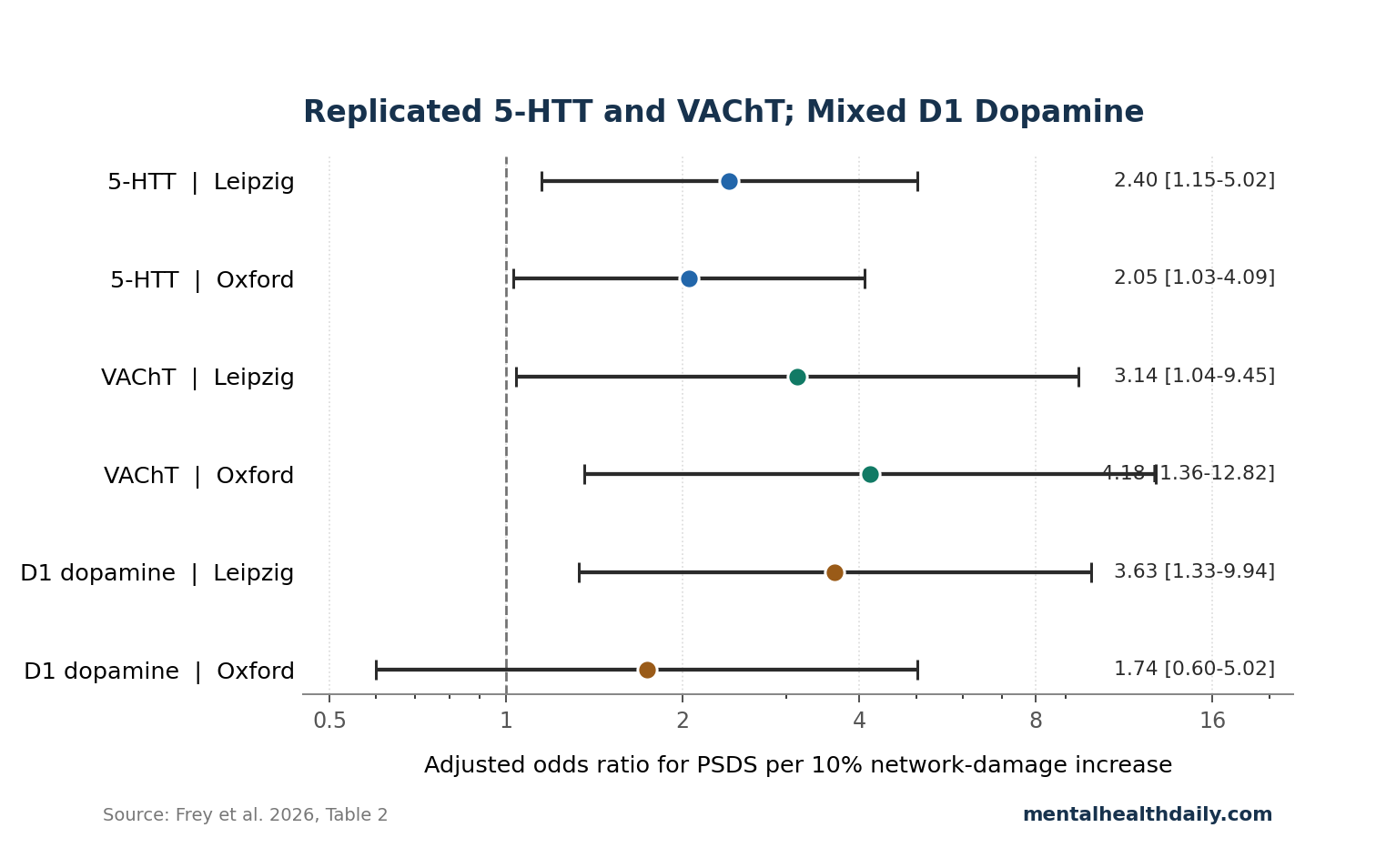
Serotonin transporter (5-HTT) is the protein targeted by selective serotonin reuptake inhibitors (SSRIs), which block serotonin reuptake and increase synaptic serotonin signaling. In Frey et al., each 10% increase in 5-HTT-weighted network damage was associated with higher odds of post-stroke depressive symptoms in Leipzig (OR 2.4, 95% CI 1.15–5.02, p = 0.020, Delta AIC 3.38) and Oxford (OR 2.05, 95% CI 1.03–4.09, p = 0.041, Delta AIC 2.27).1
Vesicular acetylcholine transporter (VAChT) packages acetylcholine into synaptic vesicles so cholinergic neurons can release it. VAChT-weighted network damage also predicted depressive symptoms in both cohorts: Leipzig OR 3.14 (95% CI 1.04–9.45, p = 0.042, Delta AIC 2.11) and Oxford OR 4.18 (95% CI 1.36–12.82, p = 0.012, Delta AIC 4.47).1
D1 dopamine damage looked strong in Leipzig (OR 3.63, 95% CI 1.33–9.94, p = 0.012), but Oxford did not reproduce it (OR 1.74, 95% CI 0.60–5.02, p = 0.306). That is not a proof that dopamine is irrelevant to post-stroke depression.
It is a warning against turning this paper into a generalized dopamine-anhedonia explanation when the replicated cohort-level signal was 5-HTT plus VAChT.1
Why VAChT Makes the Result More Than an SSRI Story
A serotonin finding in post-stroke depression is not surprising. SSRIs are common antidepressants, 5-HTT genetics have been studied in post-stroke depression susceptibility, and fluoxetine trials after stroke have tested whether broad serotonergic treatment changes recovery or depression outcomes.34
The VAChT signal is the more interesting addition because acetylcholine links mood, attention, cognitive slowing, arousal, and neuroimmune regulation. Cholinergic signaling is the acetylcholine-based communication system used by brain circuits involved in attention and memory as well as autonomic and inflammatory control.
Frey et al. did not show that cholinergic drugs treat post-stroke depression, but the VAChT result gives a concrete network reason to examine patients whose depression travels with slowed cognition, apathy, or attentional impairment.15
Fluoxetine evidence keeps the interpretation calibrated. Large post-stroke fluoxetine trials did not produce the functional-recovery breakthrough that early enthusiasm hoped for, and later individual-patient-data work found only modest depression-prevention signals alongside adverse events such as fractures and hyponatremia.4
The Frey result fits a narrower idea: serotonin-weighted lesion damage may identify one vulnerability pathway, but broad serotonergic treatment for unselected stroke patients is not automatically justified.
Network Damage Explains More Than Lesion Location
Older lesion-location theories tried to tie post-stroke depression to left frontal or other focal lesion sites. Reviews and later imaging work made that view unstable: many depressed patients had lesions outside the proposed regions, and many patients with proposed risk-region lesions did not develop depression.6
Lesion-network mapping changed the question. Padmanabhan et al. showed that focal lesions associated with depression can converge on a common functional network connected to the left dorsolateral prefrontal cortex.7
Pan et al. later used structural disconnection features to predict post-stroke depression, and Klingbeil et al. connected depressive symptoms after stroke with lesion location, structural disconnection, and functional diaschisis.89
Diaschisis means dysfunction in brain regions remote from the visible lesion because connected networks have been disrupted. Frey et al. add another layer to that network logic: a stroke may be risky more than because it disconnects a mood-related circuit, but because the disconnected circuit sits inside a serotonin- or acetylcholine-heavy architecture.1
What the ORs Can and Cannot Support
The table values are large enough to take seriously, but they are not a clinical prediction model. Frey et al. explicitly framed the analysis as mechanistic inference rather than individual-level prediction, and no out-of-sample prediction was performed.1
- Real support: across 2 cohorts, 5-HTT and VAChT network damage were associated with HADS-D-defined post-stroke depressive symptoms after adjustment for demographics, lesion volume, and neurological deficit.
- Not supported yet: the model cannot diagnose depression, select an antidepressant, prove serotonin neurons were destroyed, or show that cholinergic medication should be added after stroke.
- Main design limit: the neurotransmitter maps and structural connectomes were normative. They reflect average receptor/transporter distributions and average wiring, not patient-specific PET scans or patient-specific diffusion MRI.
- Timing limit: depression was measured at one 6-month follow-up, so the analysis cannot separate early vulnerability, treatment exposure, symptom persistence, and recovery trajectory.
That evidence-strength note is especially important because the paper is a medRxiv preprint. The result is useful as a mechanistic lead, but it should not be used as a direct guide to clinical treatment before peer review, prospective validation, and treatment-response testing.
Questions Raised by the Frey Network Study
Does this prove that stroke damages serotonin or acetylcholine neurons?
No. The study scored lesions by how much they disrupted networks weighted by PET-derived 5-HTT and VAChT maps.
That is different from showing direct loss of serotonin-producing or acetylcholine-producing neurons in each patient.1
Why did the dopamine result matter if it did not replicate?
The authors’ hypothesis specifically mentioned dopaminergic systems, and D1 damage was significant in Leipzig. Oxford failed to reproduce that association, so dopamine remains a phenotype-specific or cohort-specific lead rather than a stable marker in this dataset.1
Should patients with stroke-related 5-HTT network damage receive SSRIs?
Not from this study alone. Existing fluoxetine trials show that broad SSRI treatment after stroke has tradeoffs, including adverse-event risk, and Frey et al. did not test treatment response by 5-HTT network score.4
What would make the VAChT finding clinically useful?
Prospective studies would need to show that VAChT-weighted damage predicts depressive-symptom trajectory, cognitive-affective subtype, or response to a specific intervention. Without that step, VAChT is a strong mechanistic clue rather than a treatment-matching tool.
References
- Frey BM, Klingbeil J, Moore MJ, et al. Linking network damage to post-stroke depression: a neurotransmitter-informed connectome analysis. medRxiv. 2026. doi:10.64898/2026.04.24.26351561
- Hackett ML, Pickles K. Part I: frequency of depression after stroke: an updated systematic review and meta-analysis of observational studies. International Journal of Stroke. 2014;9(8):1017–1025. doi:10.1111/ijs.12357
- Mak KK, Kong WY, Mak A, Sharma VK, Ho RC. Polymorphisms of the serotonin transporter gene and post-stroke depression: a meta-analysis. Journal of Neurology, Neurosurgery & Psychiatry. 2013;84(3):322–328. doi:10.1136/jnnp-2012-303791
- Mead G, Graham C, Lundstrom E, et al. Individual patient data meta-analysis of the effects of fluoxetine on functional outcomes after acute stroke. International Journal of Stroke. 2024;19(7):798–808. doi:10.1177/17474930241242628
- Dagyte G, Den Boer JA, Trentani A. The cholinergic system and depression. Behavioural Brain Research. 2011;221(2):574–582. doi:10.1016/j.bbr.2010.02.023
- Robinson RG, Jorge RE, Starkstein SE. Poststroke depression: an update. The Journal of Neuropsychiatry and Clinical Neurosciences. 2024;36(1):22–35. doi:10.1176/appi.neuropsych.21090231
- Padmanabhan JL, Cooke D, Joutsa J, et al. A human depression circuit derived from focal brain lesions. Biological Psychiatry. 2019;86(10):749–758. doi:10.1016/j.biopsych.2019.07.023
- Pan C, Li G, Jing P, et al. Structural disconnection-based prediction of poststroke depression. Translational Psychiatry. 2022;12(1):461. doi:10.1038/s41398-022-02223-2
- Klingbeil J, Brandt ML, Stockert A, et al. Associations of lesion location, structural disconnection, and functional diaschisis with depressive symptoms post stroke. Frontiers in Neurology. 2023;14:1144228. doi:10.3389/fneur.2023.1144228
- Hansen JY, Shafiei G, Markello RD, et al. Mapping neurotransmitter systems to the structural and functional organization of the human neocortex. Nature Neuroscience. 2022;25(11):1569–1581. doi:10.1038/s41593-022-01186-3