
Multiple system atrophy is a rapidly progressive neurodegenerative disease with no proven disease-modifying therapy. A 2026 review by Jeong and colleagues tracks two decades of repurposed-drug trials, with one recent positive Phase 2 result.1
Research Highlights
- Multiple system atrophy (MSA) is a fatal α-synuclein disorder distinct from Parkinson’s disease. It progresses faster than PD (median survival 5–10 years from onset), responds poorly or transiently to dopaminergic therapy, and has no FDA-approved disease-modifying treatment as of 2026.2
- Drug repurposing has been the dominant therapeutic strategy. Agents already approved for other indications — sirolimus, rifampicin, lithium, nilotinib, EGCG, minocycline, IVIG, ubiquinol, riluzole, fluoxetine, exendin-4, and others — have been tested in MSA models and trials based on mechanistic overlap with α-synuclein aggregation, neuroinflammation, mitochondrial dysfunction, and neurotrophic deficits.1
- Most repurposed agents have failed in clinical trials despite preclinical promise. Rifampicin, rasagiline, minocycline, lithium, fluoxetine, intravenous immunoglobulin, and several others showed disease-relevant effects in MSA models but didn’t translate to slower progression in randomized human trials.1
- Ubiquinol (a reduced form of CoQ10) is the notable exception. A Phase 2 placebo-controlled trial in 2023 showed significant slowing of motor progression in MSA — the first placebo-controlled evidence of disease modification in this disease.3 The mechanism involves mitochondrial-electron-transport support and antioxidant activity.
- The repurposing failure pattern is systematic, not random. Late-stage patient enrollment (where neurodegenerative damage is already advanced), small sample sizes, single-target therapies in a multi-pathway disease, and tolerability problems (lithium, nilotinib) recur across negative trials. Improved trial design and biomarker development are the field’s bottleneck, not drug-candidate scarcity.
Multiple system atrophy is one of the most aggressive neurodegenerative diseases in medicine, with median survival from symptom onset of 5–10 years and progressive disability that quickly outpaces what supportive care can offset.
Pathologically, MSA is defined by misfolded α-synuclein accumulating in oligodendrocytes (the cells that produce myelin) rather than in neurons — the same protein that drives Parkinson’s disease, but in a different cell type and with substantially worse outcomes.2
Drug development for MSA has been a graveyard of promising preclinical signals that didn’t translate.
The Jeong 2026 review surveys two decades of attempts, structured around the major pathogenic pathways — α-synuclein aggregation, neuroinflammation, mitochondrial dysfunction, neurotrophic deficits — and identifies the one positive Phase 2 trial that has emerged from this otherwise discouraging literature.1
Jeong 2026: Mapping the Repurposing Landscape
The trigger paper is a structured review of disease-modifying drug-repurposing efforts in MSA, organized by mechanism class.1 The four major pathogenic pathways and the drugs tested against each:
- α-Synuclein aggregation: sirolimus (rapamycin, mTOR inhibitor that boosts autophagy), rifampicin (binds and disaggregates α-synuclein in vitro), lithium (autophagy inducer), nilotinib (tyrosine kinase inhibitor with autophagy effects), epigallocatechin gallate / EGCG (green tea polyphenol with anti-aggregation activity).
- Neuroinflammation: minocycline (tetracycline antibiotic with anti-microglial activity), intravenous immunoglobulin (broad anti-inflammatory).
- Mitochondrial dysfunction and excitotoxicity: ubiquinol (CoQ10), rasagiline (MAO-B inhibitor with claimed neuroprotective effects), safinamide (MAO-B inhibitor + sodium-channel modulator), riluzole (glutamate-release inhibitor approved for ALS).
- Neurotrophic support: fluoxetine and other SSRIs (BDNF-upregulating effects), insulin (intranasal, neuroprotective signaling), exendin-4 / GLP-1 agonists (neuroprotective via GLP-1 receptor signaling).
For each agent, the review covered preclinical mechanism evidence, animal-model results, and clinical trial outcomes where available.
The Repurposing Failure Pattern Is Consistent
The dominant story is one of preclinical promise not translating to clinical disease modification. Specific examples:1
- Rifampicin: Preclinical work showed it could disaggregate α-synuclein in vitro and reduce pathology in transgenic mouse models. The 2014 randomized trial in MSA was stopped for futility — no slowing of progression.4
- Rasagiline: Selective MAO-B inhibitor with proposed neuroprotective effects. The 2013 PROMESA trial in MSA found no effect on progression.5
- Minocycline: Broad anti-microglial agent with promising MSA-model data. Clinical trials showed no significant disease modification.6
- Intravenous immunoglobulin (IVIG): Tested based on broad anti-inflammatory rationale; small open-label trials suggested signal but placebo-controlled work didn’t replicate.7
- Lithium: Autophagy-induction rationale strong in preclinical models. Clinical trials hampered by tolerability problems — MSA patients often unable to tolerate therapeutic doses due to baseline autonomic dysfunction.1
- Fluoxetine: SSRIs broadly upregulate BDNF, providing a neurotrophic rationale. Clinical trial data mixed; no convincing disease modification.8
This is not for lack of trying. Each of these agents had a defensible mechanistic rationale and preclinical support before entering clinical testing. The systematic failure pattern points at structural problems in the development approach rather than wrong drug choices.
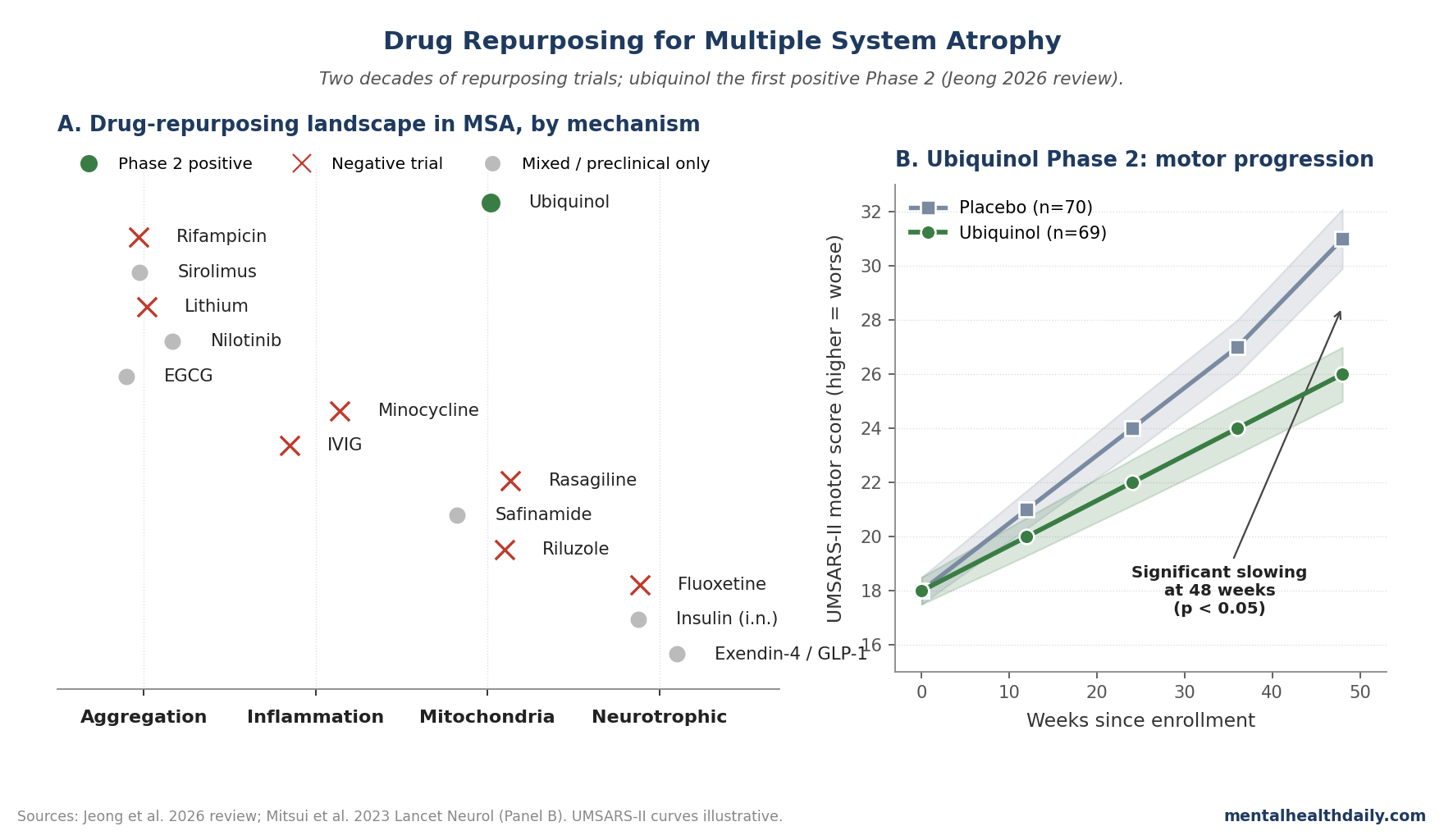
Ubiquinol: The First Positive Phase 2 Disease-Modification Signal
Ubiquinol is the reduced (active) form of coenzyme Q10. It functions as an electron carrier in the mitochondrial electron transport chain and as a lipid-soluble antioxidant. The mechanistic rationale for MSA is that α-synuclein pathology produces mitochondrial dysfunction, energy failure, and oxidative damage in oligodendrocytes and neurons; supporting mitochondrial function may slow this cascade.3
The trial Jeong highlights as the field’s first positive result is the Mitsui 2023 Phase 2 placebo-controlled trial of ubiquinol in MSA. Headline findings:3
- Significant slowing of motor progression on the Unified Multiple System Atrophy Rating Scale Part II (UMSARS-II) at 48 weeks in the ubiquinol arm vs. placebo.
- Effect size was clinically meaningful rather than statistically significant but trivially small — the magnitude of difference exceeded what would be expected from random variation.
- Tolerability was acceptable — ubiquinol’s safety profile from CoQ10 supplementation history meant no significant adverse-event signal.
This is the first placebo-controlled evidence of disease modification in MSA after roughly two decades of negative repurposing trials. The result needs Phase 3 confirmation before becoming standard care, but it represents a meaningful change in the prognostic conversation.
Three observations matter for interpreting the ubiquinol result.
- The mechanism overlap with PD is partial. Coenzyme Q10 has been tested in Parkinson’s disease and largely failed (Q10 trials in PD showed no significant disease modification). Ubiquinol’s positive result in MSA suggests either MSA-specific mitochondrial vulnerability that PD doesn’t share, or that the active reduced form (ubiquinol) is bioactive in ways that conventional CoQ10 isn’t.9
- The effect size is moderate. Ubiquinol slowed progression but didn’t halt it. MSA still progresses on ubiquinol; the slowing is incremental rather than transformative. This is consistent with how disease-modifying therapies in adjacent neurodegenerative diseases (anti-amyloid drugs in Alzheimer’s, GLP-1 agonists in Parkinson’s) typically show: meaningful but modest slowing rather than disease arrest.
- Phase 3 confirmation isn’t a formality. Phase 2 positive results in neurodegenerative drug development have a meaningful failure rate at Phase 3 (the most prominent recent example being simufilam in Alzheimer’s). Whether ubiquinol’s effect replicates in larger, more diverse, longer-duration trials remains genuinely uncertain.
Why Most MSA Repurposing Trials Have Failed
The pattern is structural. Three drivers recur across negative trials:
- Late-stage enrollment. By the time a patient is diagnosed with MSA, oligodendrocyte loss and downstream neurodegeneration are typically already advanced. Disease-modifying interventions in advanced neurodegeneration have repeatedly failed across diseases (anti-amyloid drugs in symptomatic AD, anti-tau in PSP). Earlier intervention requires earlier diagnosis, which in MSA requires biomarkers that are still in development.
- Single-target therapies in a multi-pathway disease. α-Synuclein aggregation, neuroinflammation, mitochondrial dysfunction, and neurotrophic deficits aren’t independent — they form a self-reinforcing cascade. Targeting any single pathway with a moderately potent agent may produce minimal slowing if the other pathways continue. Combination therapies, while logistically complex, are increasingly viewed as necessary for effective MSA disease modification.1
- Small sample sizes and short follow-up. MSA’s rarity (prevalence around 3–4 per 100,000) makes large trials hard to enroll. Most published negative trials have had sample sizes of 50–200 and follow-up windows of 6–12 months — durations that may be too short to detect clinically meaningful disease-modification signals against the backdrop of MSA’s already rapid progression.
The ubiquinol trial succeeded partly by being one of the larger, longer Phase 2 trials in MSA, with biomarker-supported endpoints. This template (larger samples, longer follow-up, biomarker-anchored endpoints) is what successful future MSA trials likely require.
What Popular Coverage of Neurodegenerative Drug Development Misses
Three calibrations matter when reading coverage of MSA and related diseases.
- “No treatment for MSA” oversells the symptomatic-care gap. While disease-modifying therapy is unproven, symptomatic management of orthostatic hypotension (midodrine, fludrocortisone), urinary symptoms, and motor symptoms (transient response to levodopa in some patients) provides meaningful quality-of-life support. The disease-modification gap is real but doesn’t mean nothing can be done.
- Repurposing isn’t a shortcut. The “drug already approved, so faster to market” framing oversimplifies. Repurposed drugs still require new RCTs, regulatory review for new indications, and dose-finding work. The repurposing-development timeline is faster than de novo drug development but is still 5–10 years from initial promising signal to approved indication.
- Ubiquinol’s positive result doesn’t mean CoQ10 supplements help MSA. The Phase 2 trial used a specific high-dose ubiquinol formulation under monitored conditions. Standard over-the-counter CoQ10 supplements (typically the oxidized ubiquinone form, often at lower doses) haven’t been tested under the same rigor and shouldn’t be assumed to produce equivalent effects. Patients with MSA considering supplementation should discuss with treating neurologists, not self-substitute.
Limitations of the Jeong Synthesis
Review-level analysis, not meta-analysis. The Jeong paper is a structured narrative review summarizing trial outcomes by mechanism class, not a quantitative meta-analysis with pooled effect sizes. The qualitative conclusions are appropriate but don’t allow the precision of meta-analytic estimates.
Publication bias affects the negative-trial picture. Some trials with neutral results may not have been published, while the more dramatic negative results (rifampicin futility termination) are well-known. The actual repurposing failure rate may be slightly different from what the published literature shows.
The ubiquinol Phase 2 result is single-trial evidence. One positive trial doesn’t establish a treatment standard. Phase 3 confirmation, replication in different populations, and longer-duration follow-up are all needed before ubiquinol becomes part of standard MSA care.
Combination-therapy approaches are the obvious next step but largely untested. Combining mitochondrial support (ubiquinol) with anti-aggregation (rapamycin or similar) with neuroinflammation modulation (minocycline or similar) makes mechanistic sense, but the trial architecture for combination-therapy testing in rare-disease populations is challenging and largely undeveloped.
MSA subtype heterogeneity isn’t fully addressed. MSA-P (parkinsonian-predominant) and MSA-C (cerebellar-predominant) likely have different molecular trajectories and different drug responsiveness. Most reviewed trials have pooled both subtypes; subtype-specific results may differ from the pooled picture.
Biomarker-supported trial endpoints remain underdeveloped. Reliable in-vivo α-synuclein quantification, oligodendrocyte-specific imaging, and disease-progression biomarkers would dramatically improve trial efficiency in MSA. The seed amplification assay (SAA) is the most promising biomarker development of the past 5 years and is starting to enter trial design, but standardization and clinical validation are ongoing.10
Practical Implications for Patients and Families
For patients with MSA and their families, three practical takeaways follow.
- Symptomatic management remains the priority. Orthostatic hypotension management (midodrine, fludrocortisone, droxidopa, postural training, increased fluid/salt intake), urinary symptom management, dysphagia management, and supportive physical and occupational therapy meaningfully improve quality of life regardless of disease-modifying therapy availability. Specialty MSA centers (in the US, the MSA Coalition Center of Excellence network) provide more comprehensive symptomatic care than community neurology.
- Clinical trial enrollment is reasonable to consider. The repurposing literature has been disappointing, but the field is actively testing newer agents including ubiquinol confirmation studies, novel anti-α-synuclein immunotherapies, and combination approaches. Early-stage MSA patients are the population most likely to benefit from any successful disease-modifying agent. ClinicalTrials.gov maintains current listings.
- Ubiquinol-as-prescription discussion should happen with the treating neurologist. The Phase 2 positive result is meaningful but not yet standard of care. Some MSA specialists have begun off-label prescribing of high-dose ubiquinol based on the Phase 2 data plus the favorable safety profile. Whether this is appropriate for any individual patient depends on clinical context, comorbidities, and the local practice landscape. Discussion with a movement-disorders specialist familiar with the literature is the right path.
Questions About MSA Treatment
What is multiple system atrophy?
A progressive neurodegenerative disease characterized by misfolded α-synuclein accumulating in oligodendrocytes (the cells that produce myelin), with downstream neuronal loss in striatonigral and olivopontocerebellar circuits. Clinically presents with combinations of parkinsonism, cerebellar ataxia, and autonomic failure (orthostatic hypotension, urinary dysfunction). Median survival from symptom onset is 5–10 years.2
Is MSA the same as Parkinson’s disease?
Related but distinct. Both involve α-synuclein pathology, but MSA accumulates α-synuclein in oligodendrocytes (forming glial cytoplasmic inclusions) while Parkinson’s accumulates it in neurons (forming Lewy bodies). MSA progresses much faster than PD, responds poorly or transiently to dopaminergic medications, and has a different clinical presentation. The two diseases are increasingly understood as related synucleinopathies with different cellular vulnerabilities.
Is there any treatment that slows MSA?
No FDA-approved disease-modifying treatment as of 2026. The 2023 Phase 2 ubiquinol trial showed significant slowing of motor progression vs. placebo — the first positive disease-modification signal in MSA — but Phase 3 confirmation is pending.3 Symptomatic management of orthostatic hypotension, urinary symptoms, and motor symptoms remains the standard of care.
Should I take CoQ10 supplements for MSA?
A reasonable question for the treating neurologist. The Phase 2 ubiquinol trial used a specific high-dose formulation; over-the-counter CoQ10 typically uses the oxidized ubiquinone form at lower doses and hasn’t been tested under the same rigor.3 Some MSA specialists are prescribing high-dose ubiquinol off-label based on the Phase 2 evidence; this isn’t yet standard practice, and self-substitution with retail supplements isn’t supported by the trial data.
Why have so many MSA drug trials failed?
Three structural reasons: (1) patients are typically enrolled at late-stage disease, where neurodegeneration is already advanced and harder to slow; (2) single-target therapies are tested in a disease driven by multiple interacting pathways; (3) sample sizes are small and follow-up durations are short due to the disease’s rarity. Combination therapies, earlier intervention via biomarkers, and longer trials are the field’s directional next steps.1
What’s the most promising experimental treatment?
Ubiquinol Phase 3 confirmation is the most clinically immediate. Beyond that, anti-α-synuclein immunotherapies (similar to anti-amyloid antibodies in Alzheimer’s) are entering clinical testing, and small molecules targeting α-synuclein aggregation are in development. The field is more active than the disappointing repurposing record suggests.
How can MSA patients access clinical trials?
Through specialty movement-disorders centers, the MSA Coalition (US) and similar advocacy organizations internationally, and ClinicalTrials.gov listings. Early-stage MSA patients (within 2–3 years of symptom onset) are the population most often eligible for disease-modification trials. Comprehensive specialty care at an MSA Center of Excellence typically includes trial-enrollment guidance.
References
- Drug repurposing for disease-modifying effects in multiple system atrophy. Jeong SH, Shin JY, Lee PH. Translational Neurodegeneration. 2026;15:15. doi:10.1186/s40035-026-00551-7
- Multiple system atrophy: state of the art. Stankovic I et al. Movement Disorders. 2019;34(11):1632-1645. doi:10.1002/mds.27897
- Efficacy and safety of high-dose ubiquinol supplementation in patients with multiple system atrophy: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Mitsui J et al. The Lancet Neurology. 2023;22(10):925-934.
- Rifampicin for the treatment of multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Low PA et al. The Lancet Neurology. 2014;13(3):268-275. doi:10.1016/S1474-4422(13)70301-6
- Rasagiline for the treatment of multiple system atrophy: a randomised, double-blind, placebo-controlled, multicentre trial. Poewe W et al. The Lancet Neurology. 2015;14(2):145-152. doi:10.1016/S1474-4422(14)70288-1
- Minocycline 1-year therapy in multiple system atrophy: effects on PET measurements of microglial activation. Dodel R et al. Movement Disorders. 2010;25(1):97-107. doi:10.1002/mds.22732
- Open-label trial of intravenous immunoglobulin (IVIG) in multiple system atrophy. Novak P et al. Acta Neurologica Scandinavica. 2012;125(4):302-307. doi:10.1111/j.1600-0404.2011.01521.x
- Effect of fluoxetine in multiple system atrophy. Friess E et al. Movement Disorders. 2006;21(11):1882-1887. doi:10.1002/mds.21072
- Coenzyme Q10 in Parkinson’s disease: a systematic review and meta-analysis. Liu J et al. European Neurology. 2011;66(1):8-16. doi:10.1159/000327370
- Discrimination of multiple system atrophy from Parkinson’s disease using α-synuclein seed amplification assay. Bargar C et al. Movement Disorders. 2024;39(7):1101-1110. doi:10.1002/mds.29796